Wang Huanzhong, Yang Jung Hyun, Chen Fang, Torres-Jerez Ivone, Tang Yuhong, Wang Mingyi, Du Qian, Cheng Xiaofei, Wen Jiangqi, Dixon Richard
Department of Plant Science and Landscape Architecture, University of Connecticut, 1390 Storrs Rd., Storrs, CT, 06269, USA.
Plant Biology Division, Samuel Roberts Noble Foundation, 2510 Sam Noble Parkway, Ardmore, OK, 73401, USA.
BMC Genomics. 2016 Jan 5;17:23. doi: 10.1186/s12864-015-2330-6.
Legumes are important to humans by providing food, feed and raw materials for industrial utilizations. Some legumes, such as alfalfa, are potential bioenergy crops due to their high biomass productivity. Global transcriptional profiling has been successfully used to identify genes and regulatory pathways in secondary cell wall thickening in Arabidopsis, but such transcriptome data is lacking in legumes.
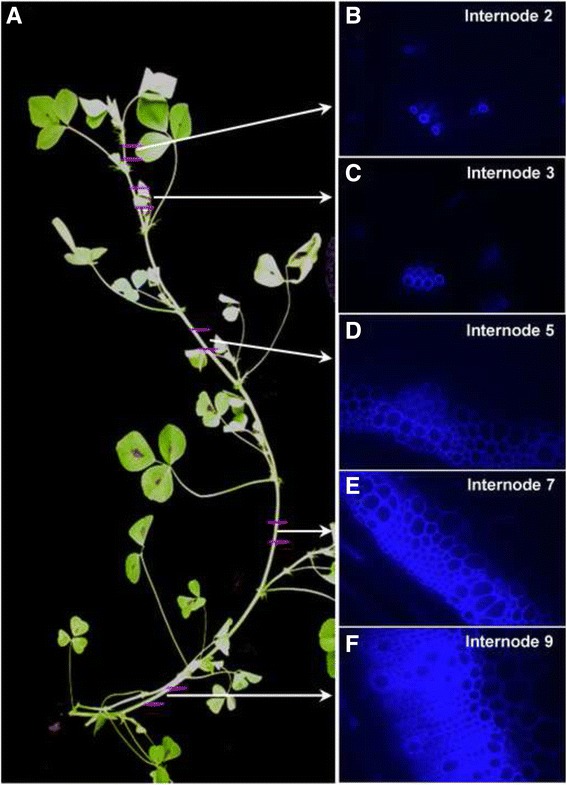
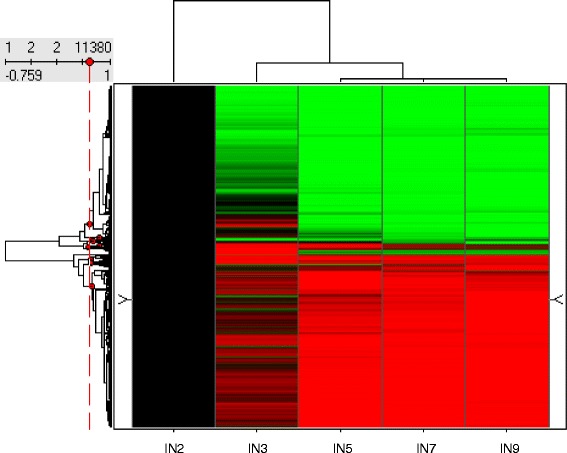
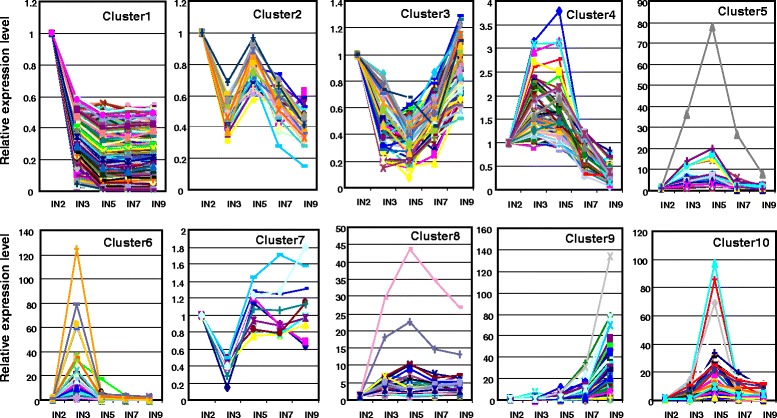
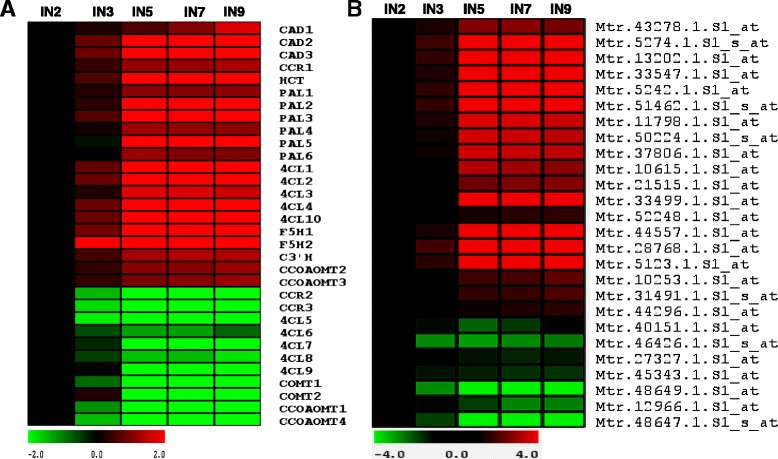
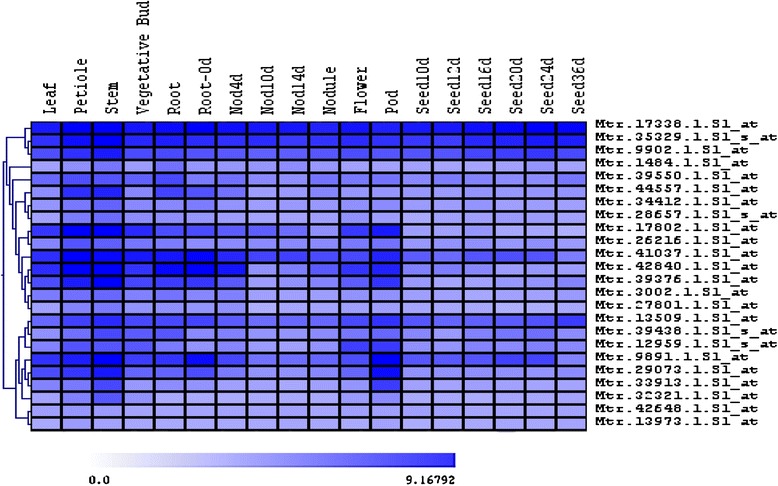
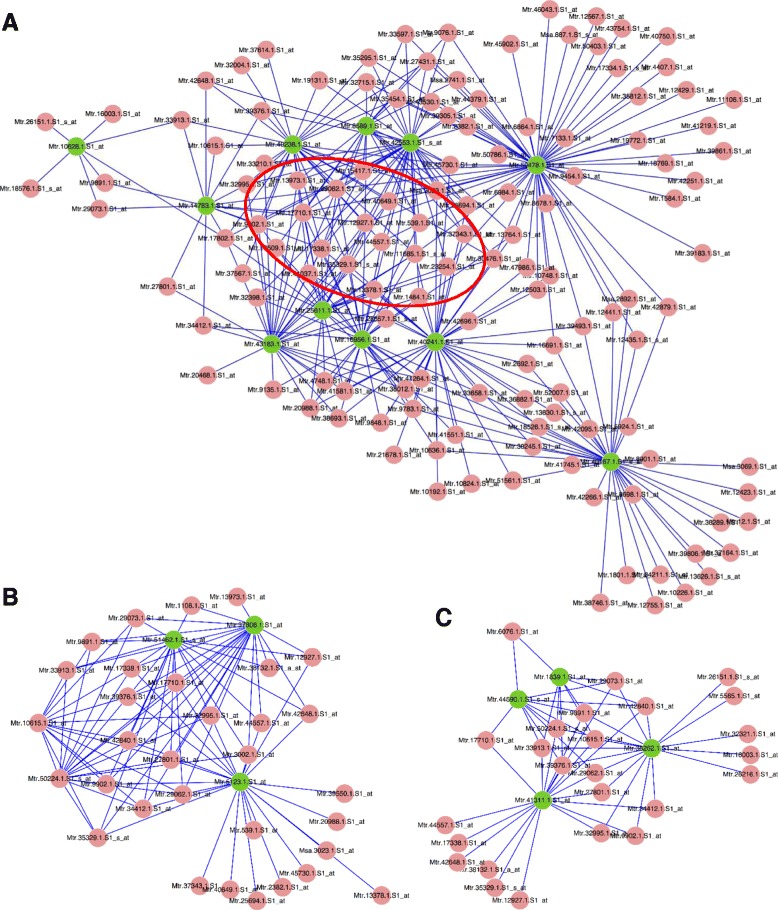
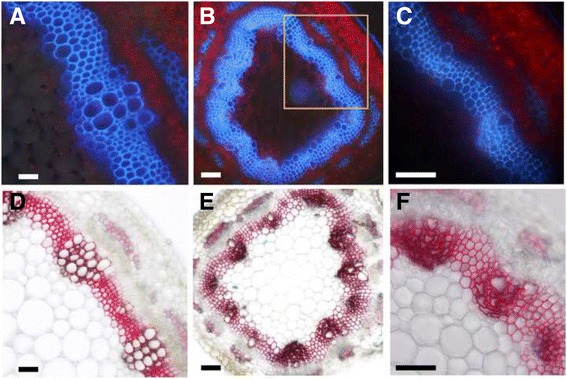
A systematic microarray assay and high through-put real time PCR analysis of secondary cell wall development were performed along stem maturation in Medicago truncatula. More than 11,000 genes were differentially expressed during stem maturation, and were categorized into 10 expression clusters. Among these, 279 transcription factor genes were correlated with lignin/cellulose biosynthesis, therefore representing putative regulators of secondary wall development. The b-ZIP, NAC, WRKY, C2H2 zinc finger (ZF), homeobox, and HSF gene families were over-represented. Gene co-expression network analysis was employed to identify transcription factors that may regulate the biosynthesis of lignin, cellulose and hemicellulose. As a complementary approach to microarray, real-time PCR analysis was used to characterize the expression of 1,045 transcription factors in the stem samples, and 64 of these were upregulated more than 5-fold during stem maturation. Reverse genetics characterization of a cellulose synthase gene in cluster 10 confirmed its function in xylem development.
This study provides a useful transcriptome and expression resource for understanding cell wall development, which is pivotal to enhance biomass production in legumes.
豆类对人类很重要,可提供食物、饲料以及工业利用的原材料。一些豆类,如苜蓿,因其高生物量生产力而成为潜在的生物能源作物。全球转录谱分析已成功用于鉴定拟南芥次生细胞壁加厚过程中的基因和调控途径,但豆类缺乏此类转录组数据。
对蒺藜苜蓿茎成熟过程中的次生细胞壁发育进行了系统的微阵列分析和高通量实时PCR分析。在茎成熟过程中有超过11000个基因差异表达,并被分为10个表达簇。其中,279个转录因子基因与木质素/纤维素生物合成相关,因此代表次生壁发育的假定调节因子。b-ZIP、NAC、WRKY、C2H2锌指(ZF)、同源异型盒和热休克因子(HSF)基因家族的表达量过高。采用基因共表达网络分析来鉴定可能调节木质素、纤维素和半纤维素生物合成的转录因子。作为微阵列的补充方法,实时PCR分析用于表征茎样本中1045个转录因子的表达,其中64个在茎成熟过程中上调超过5倍。对第10簇中的一个纤维素合酶基因进行反向遗传学表征,证实了其在木质部发育中的功能。
本研究为理解细胞壁发育提供了有用的转录组和表达资源,这对于提高豆类生物量产量至关重要。