Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA.
David H. Koch Institute for Integrative Cancer Research at MIT, Cambridge, MA 02139, USA.
Cell Rep. 2016 Jan 12;14(2):310-9. doi: 10.1016/j.celrep.2015.12.031. Epub 2015 Dec 31.
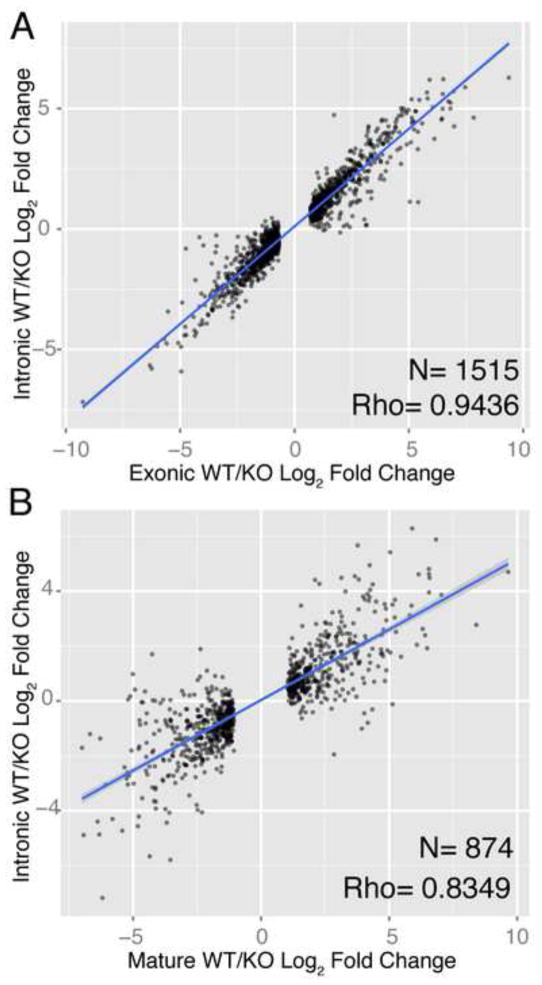
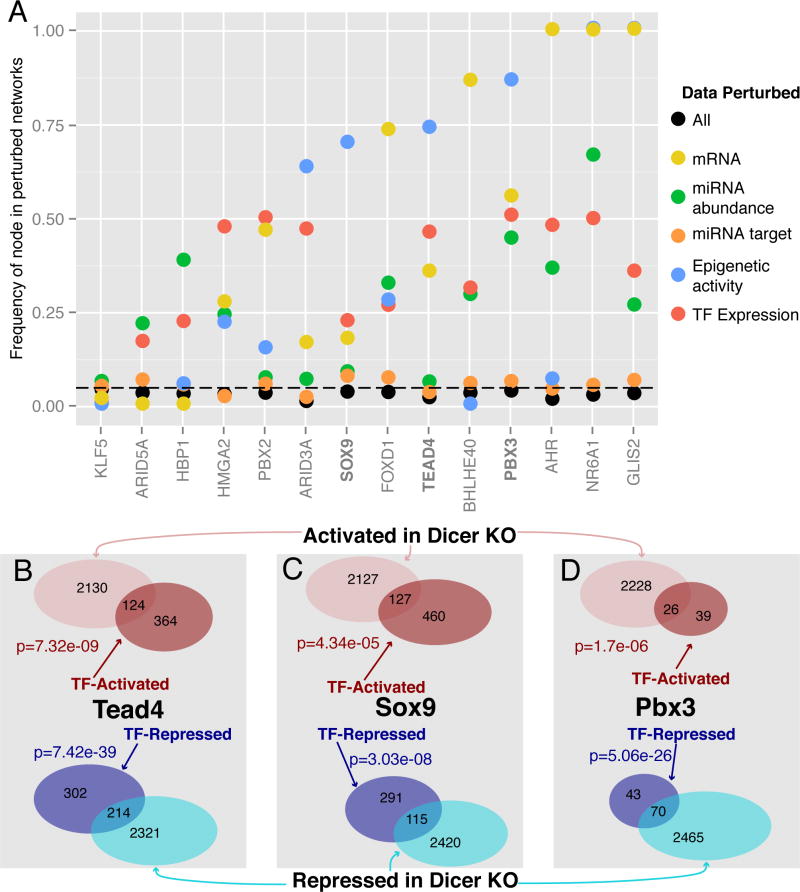
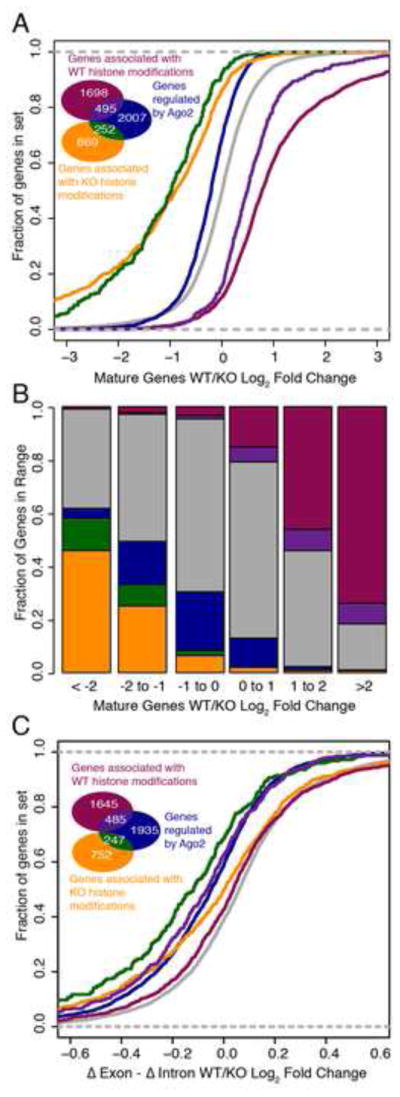
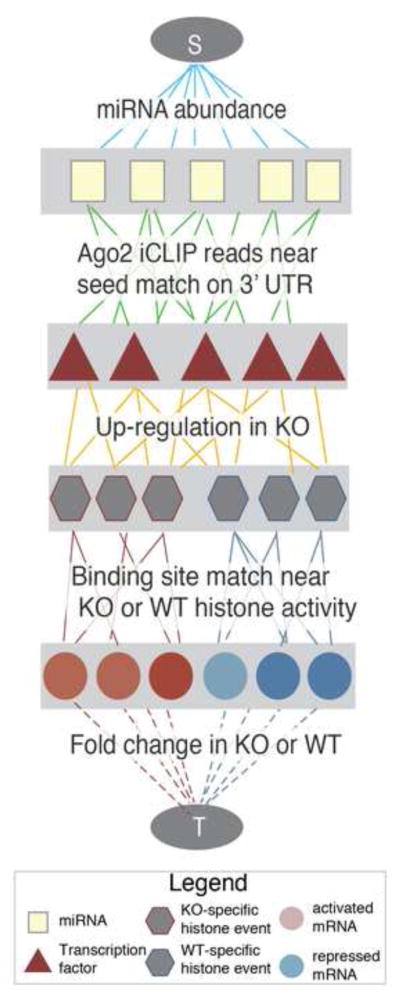
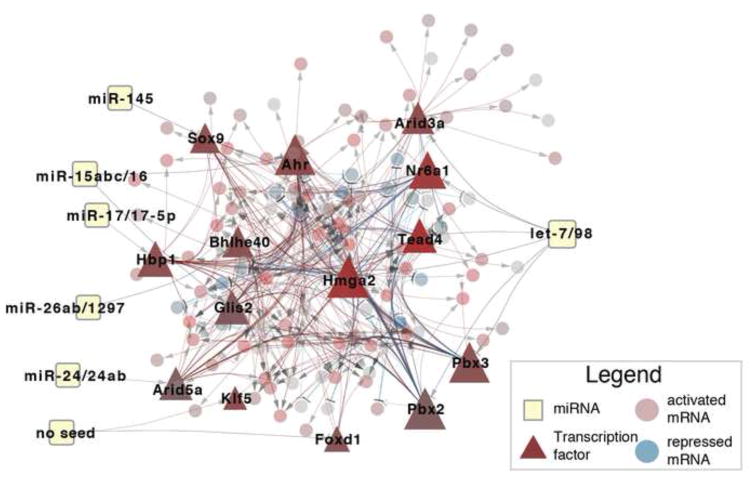
MicroRNAs (miRNAs) regulate diverse biological processes by repressing mRNAs, but their modest effects on direct targets, together with their participation in larger regulatory networks, make it challenging to delineate miRNA-mediated effects. Here, we describe an approach to characterizing miRNA-regulatory networks by systematically profiling transcriptional, post-transcriptional and epigenetic activity in a pair of isogenic murine fibroblast cell lines with and without Dicer expression. By RNA sequencing (RNA-seq) and CLIP (crosslinking followed by immunoprecipitation) sequencing (CLIP-seq), we found that most of the changes induced by global miRNA loss occur at the level of transcription. We then introduced a network modeling approach that integrated these data with epigenetic data to identify specific miRNA-regulated transcription factors that explain the impact of miRNA perturbation on gene expression. In total, we demonstrate that combining multiple genome-wide datasets spanning diverse regulatory modes enables accurate delineation of the downstream miRNA-regulated transcriptional network and establishes a model for studying similar networks in other systems.
微小 RNA(miRNAs)通过抑制 mRNA 来调节多种生物过程,但它们对直接靶标的影响较小,加上它们参与更大的调控网络,使得难以描绘 miRNA 介导的作用。在这里,我们描述了一种通过系统分析具有和不具有 Dicer 表达的一对同基因鼠成纤维细胞系中的转录、转录后和表观遗传活性来描绘 miRNA 调控网络的方法。通过 RNA 测序(RNA-seq)和交联免疫沉淀测序(CLIP-seq),我们发现大多数由全局 miRNA 缺失引起的变化都发生在转录水平。然后,我们引入了一种网络建模方法,将这些数据与表观遗传数据相结合,以鉴定特定的 miRNA 调控转录因子,这些转录因子解释了 miRNA 扰动对基因表达的影响。总的来说,我们证明了结合多个跨越不同调控模式的全基因组数据集可以准确描绘下游 miRNA 调控的转录网络,并为在其他系统中研究类似网络建立了模型。