Love Michael I, Huber Wolfgang, Anders Simon
Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8.
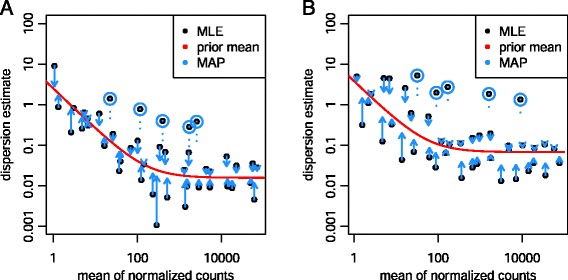
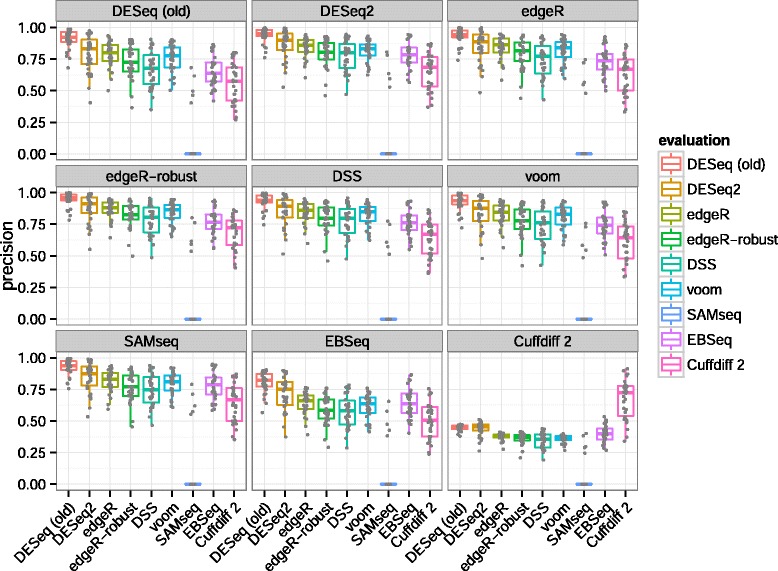
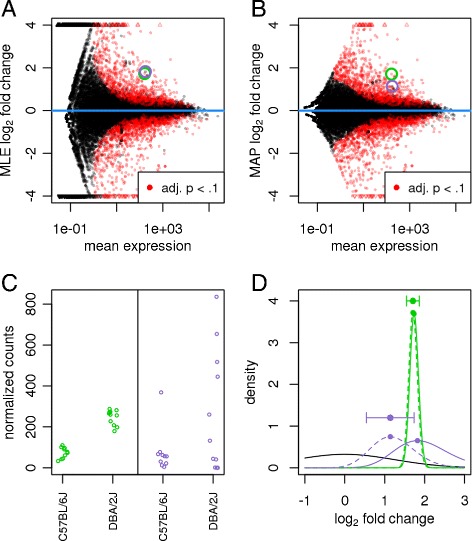
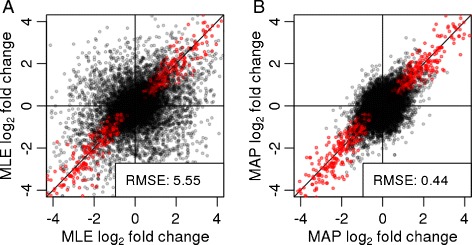
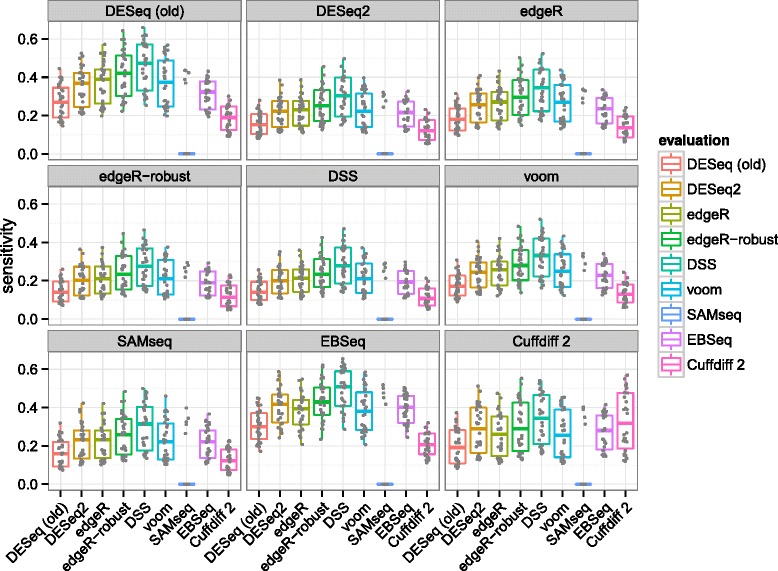
In comparative high-throughput sequencing assays, a fundamental task is the analysis of count data, such as read counts per gene in RNA-seq, for evidence of systematic changes across experimental conditions. Small replicate numbers, discreteness, large dynamic range and the presence of outliers require a suitable statistical approach. We present DESeq2, a method for differential analysis of count data, using shrinkage estimation for dispersions and fold changes to improve stability and interpretability of estimates. This enables a more quantitative analysis focused on the strength rather than the mere presence of differential expression. The DESeq2 package is available at http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html webcite.
在比较高通量测序分析中,一个基本任务是对计数数据进行分析,例如RNA测序中每个基因的读数计数,以寻找跨实验条件的系统性变化的证据。样本重复数少、离散性、动态范围大以及存在异常值等情况需要一种合适的统计方法。我们提出了DESeq2,这是一种用于计数数据差异分析的方法,它使用离散估计来估计离散度和倍数变化,以提高估计的稳定性和可解释性。这使得能够进行更定量的分析,重点关注差异表达的强度而非仅仅是差异表达的存在。DESeq2软件包可从网页http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html获取。