Bustamante Juan P, Radusky Leandro, Boechi Leonardo, Estrin Darío A, Ten Have Arjen, Martí Marcelo A
Departamento de Química Inorgánica, Analítica y Química Física, INQUIMAE-CONICET, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Buenos Aires, Argentina.
Departamento de Química Biológica e Instituto de Química Biológica de la Facultad de Ciencias Exactas y Naturales (IQUIBICEN), Universidad de Buenos Aires, Buenos Aires, Argentina.
PLoS Comput Biol. 2016 Jan 20;12(1):e1004701. doi: 10.1371/journal.pcbi.1004701. eCollection 2016 Jan.
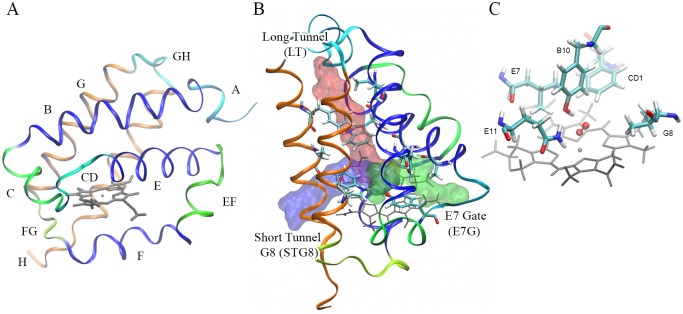
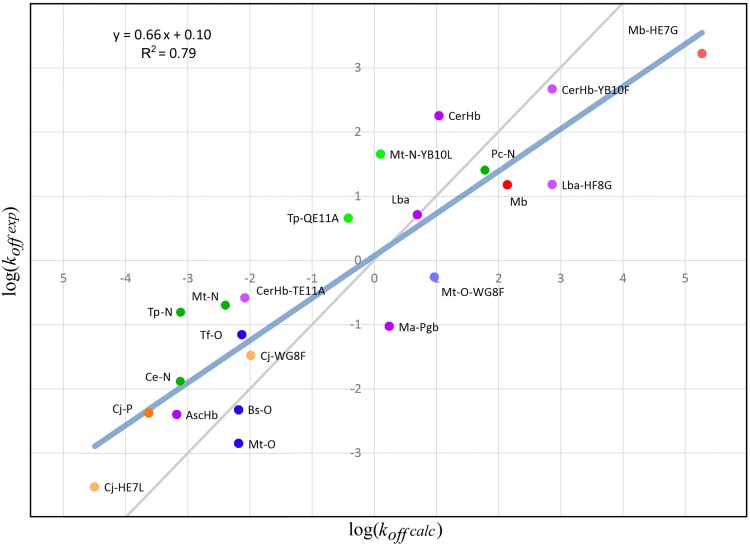
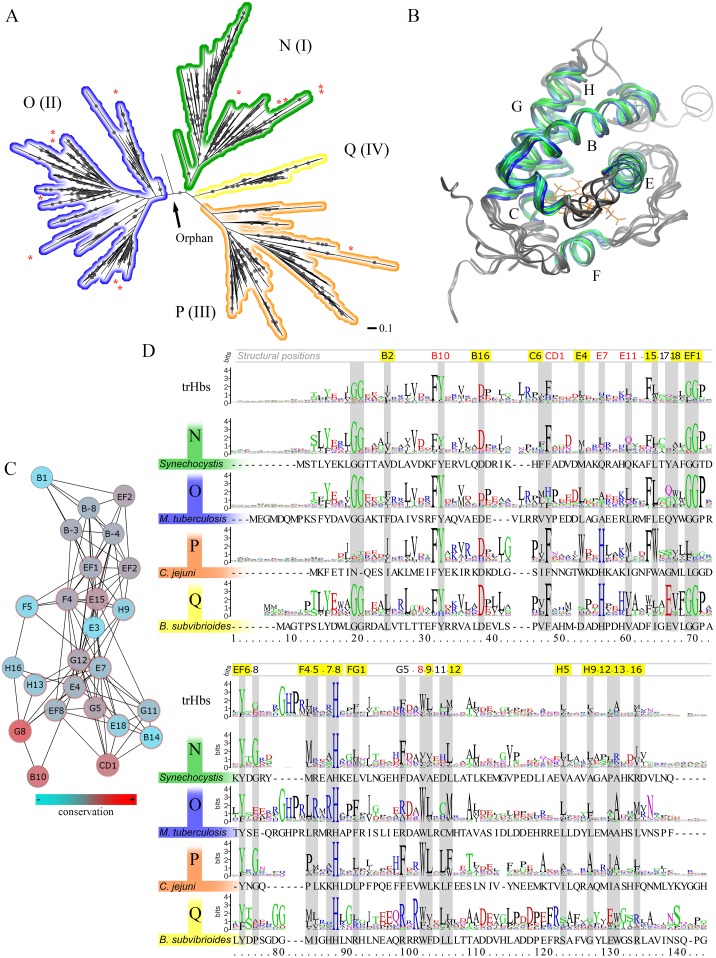
Predicting function from sequence is an important goal in current biological research, and although, broad functional assignment is possible when a protein is assigned to a family, predicting functional specificity with accuracy is not straightforward. If function is provided by key structural properties and the relevant properties can be computed using the sequence as the starting point, it should in principle be possible to predict function in detail. The truncated hemoglobin family presents an interesting benchmark study due to their ubiquity, sequence diversity in the context of a conserved fold and the number of characterized members. Their functions are tightly related to O2 affinity and reactivity, as determined by the association and dissociation rate constants, both of which can be predicted and analyzed using in-silico based tools. In the present work we have applied a strategy, which combines homology modeling with molecular based energy calculations, to predict and analyze function of all known truncated hemoglobins in an evolutionary context. Our results show that truncated hemoglobins present conserved family features, but that its structure is flexible enough to allow the switch from high to low affinity in a few evolutionary steps. Most proteins display moderate to high oxygen affinities and multiple ligand migration paths, which, besides some minor trends, show heterogeneous distributions throughout the phylogenetic tree, again suggesting fast functional adaptation. Our data not only deepens our comprehension of the structural basis governing ligand affinity, but they also highlight some interesting functional evolutionary trends.
从序列预测功能是当前生物学研究的一个重要目标,尽管当一个蛋白质被归入某个家族时可以进行宽泛的功能分配,但准确预测功能特异性并非易事。如果功能由关键结构特性决定,且相关特性可以以序列为起点进行计算,那么原则上应该能够详细预测功能。截短血红蛋白家族由于其广泛存在、在保守折叠背景下的序列多样性以及已表征成员的数量,提供了一个有趣的基准研究。它们的功能与氧气亲和力和反应性密切相关,这由缔合和解离速率常数决定,而这两者都可以使用基于计算机模拟的工具进行预测和分析。在本工作中,我们应用了一种将同源建模与基于分子的能量计算相结合的策略,在进化背景下预测和分析所有已知截短血红蛋白的功能。我们的结果表明,截短血红蛋白呈现出保守的家族特征,但其结构足够灵活,能够在几个进化步骤中实现从高亲和力到低亲和力的转变。大多数蛋白质表现出中度到高度的氧气亲和力以及多条配体迁移路径,除了一些小趋势外,这些在整个系统发育树中呈现出异质分布,再次表明功能的快速适应性。我们的数据不仅加深了我们对控制配体亲和力的结构基础的理解,还突出了一些有趣的功能进化趋势。