Koyani Chintan N, Kitz Kerstin, Rossmann Christine, Bernhart Eva, Huber Evelyn, Trummer Christopher, Windischhofer Werner, Sattler Wolfgang, Malle Ernst
Institute of Molecular Biology and Biochemistry, Medical University of Graz, Graz, Austria.
Institute of Molecular Biology and Biochemistry, Medical University of Graz, Graz, Austria; Department of Pediatrics and Adolescence Medicine, Research Unit of Osteological Research and Analytical Mass Spectrometry, Medical University of Graz, Graz, Austria.
Biochem Pharmacol. 2016 Mar 15;104:29-41. doi: 10.1016/j.bcp.2016.01.011. Epub 2016 Jan 19.
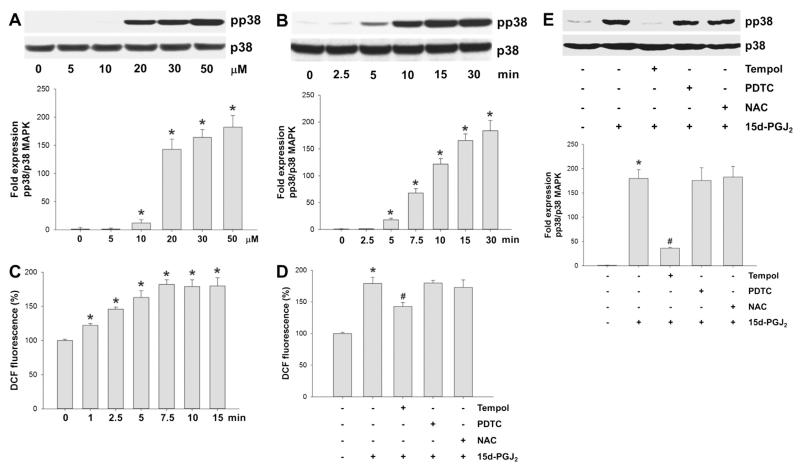
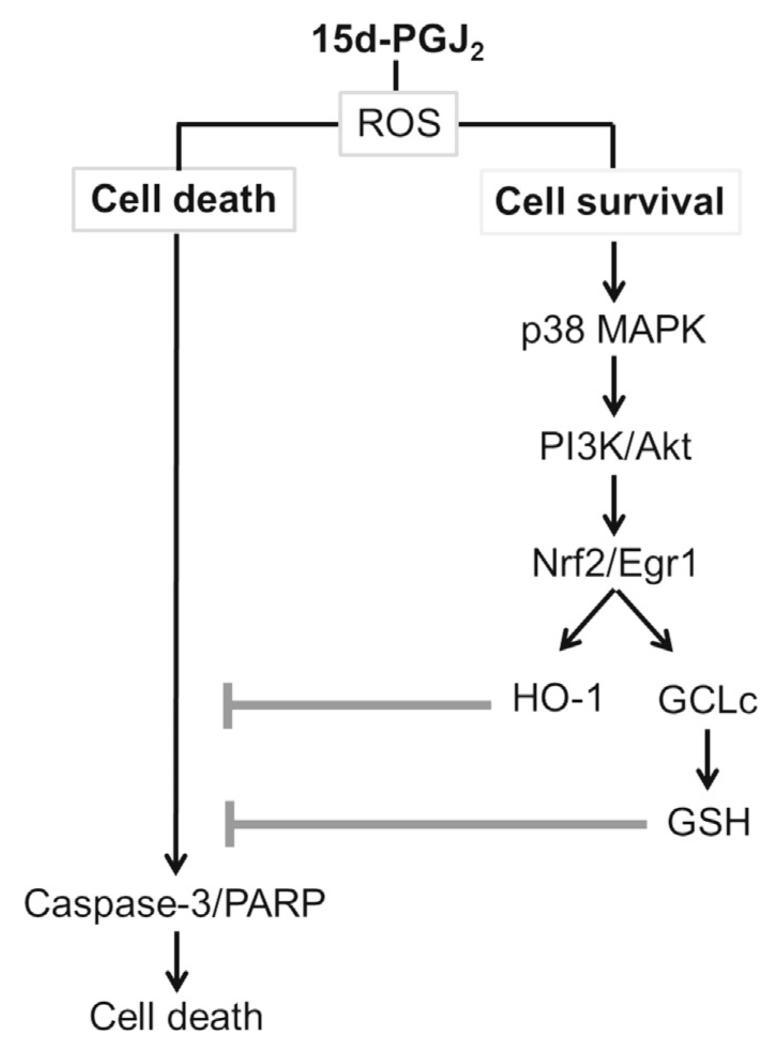
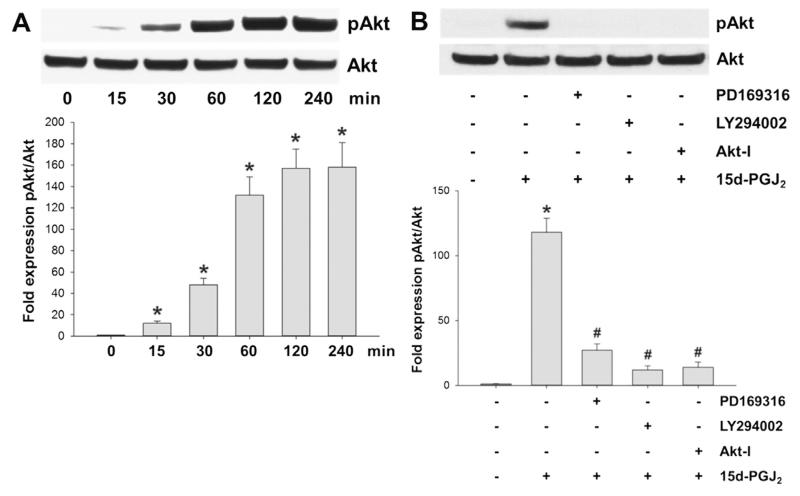
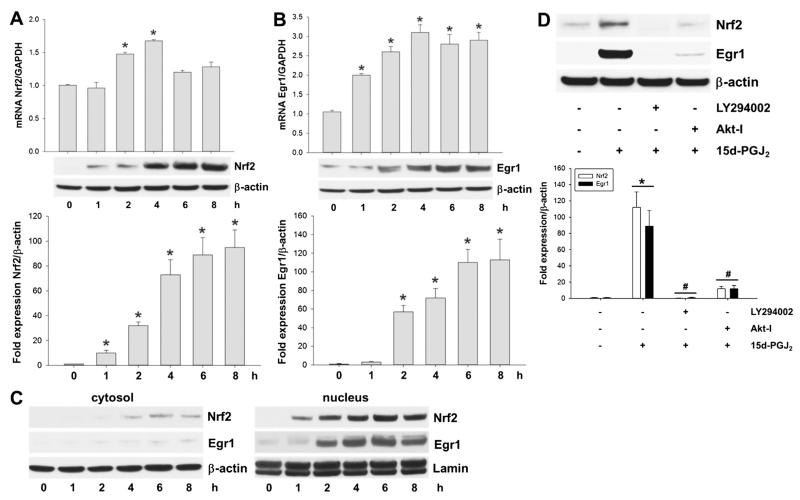
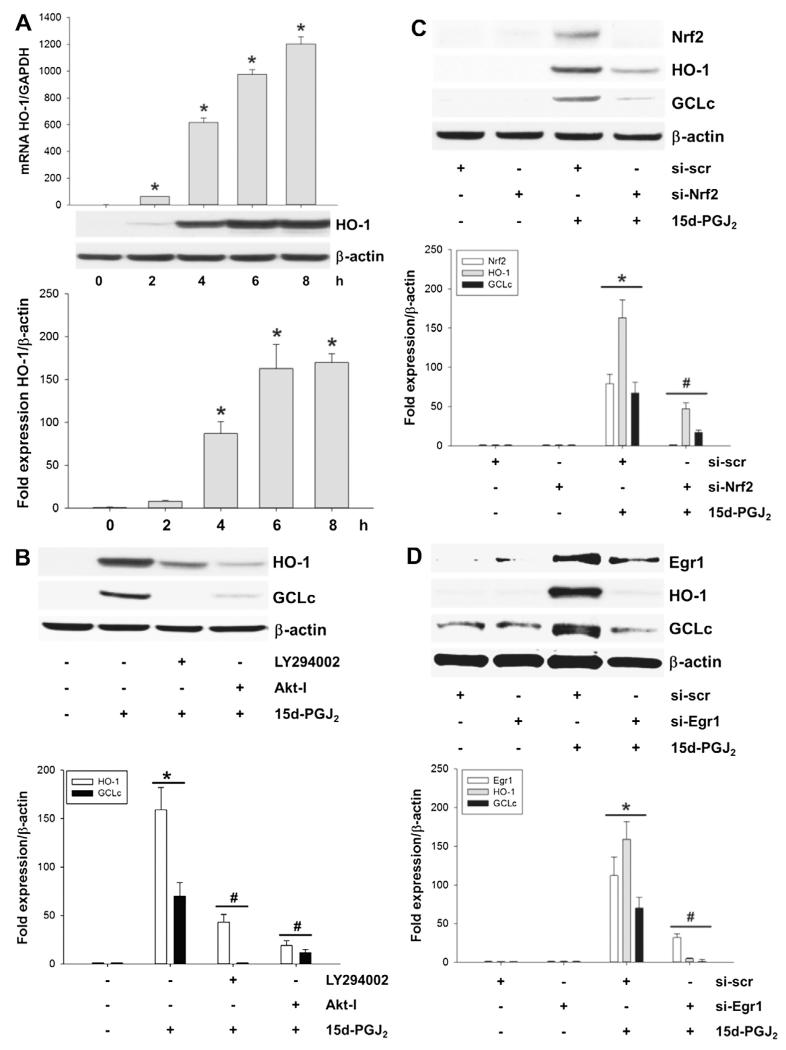
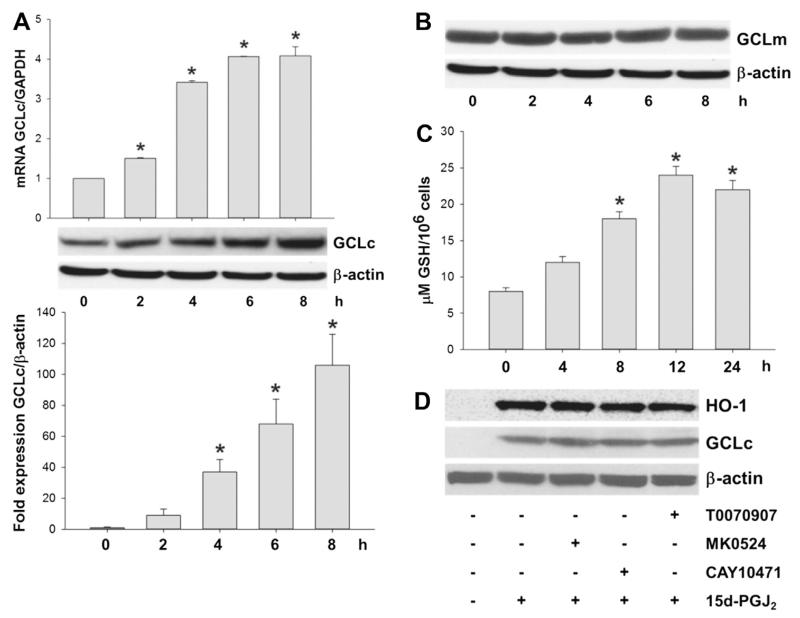
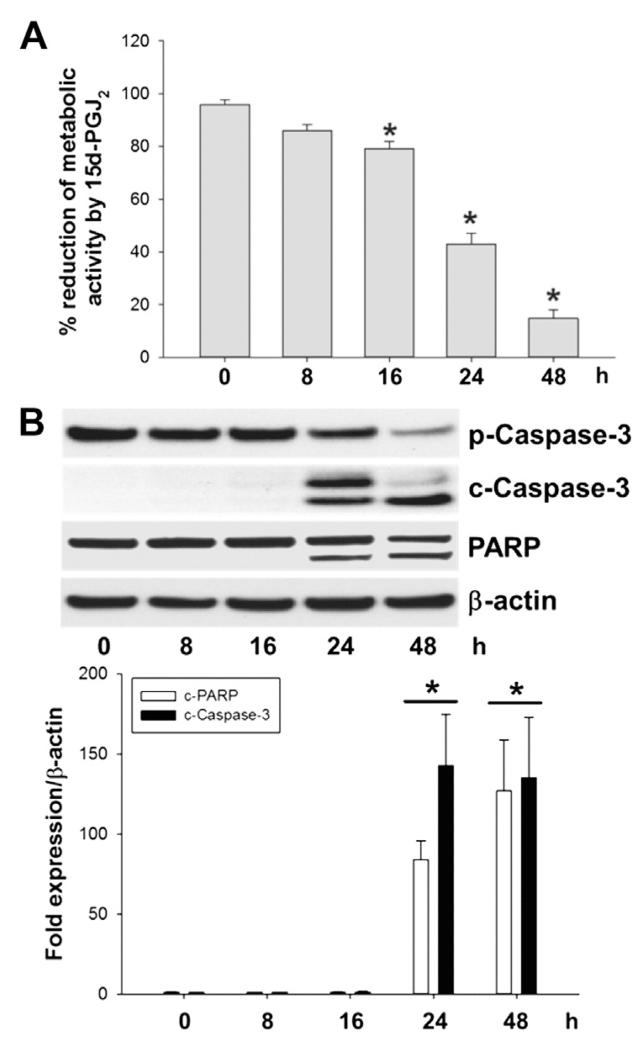
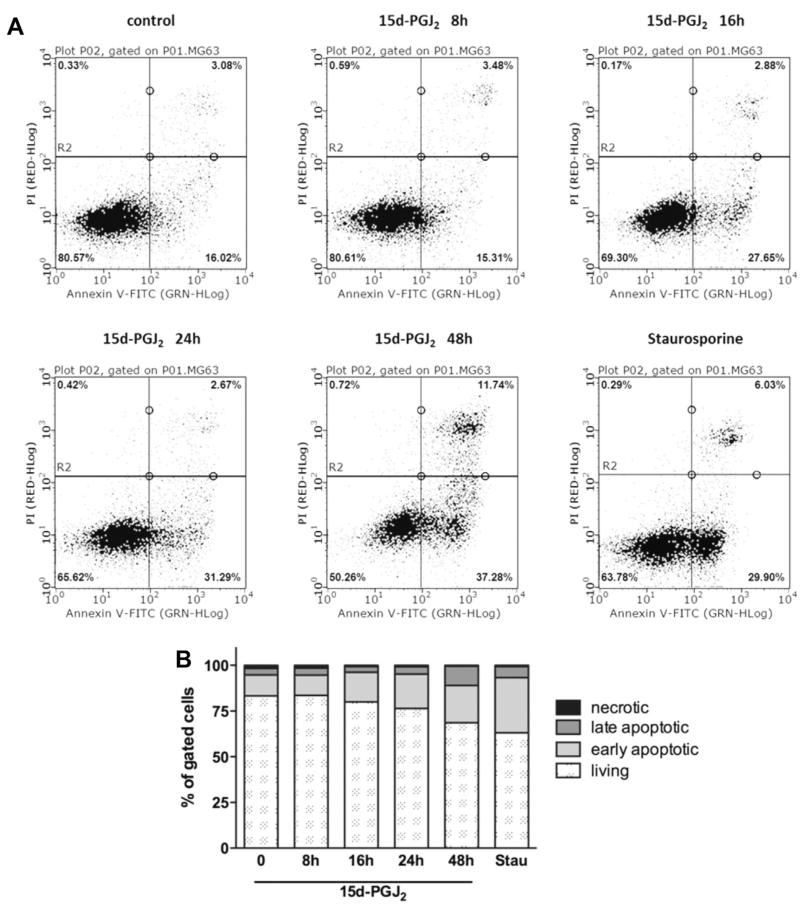
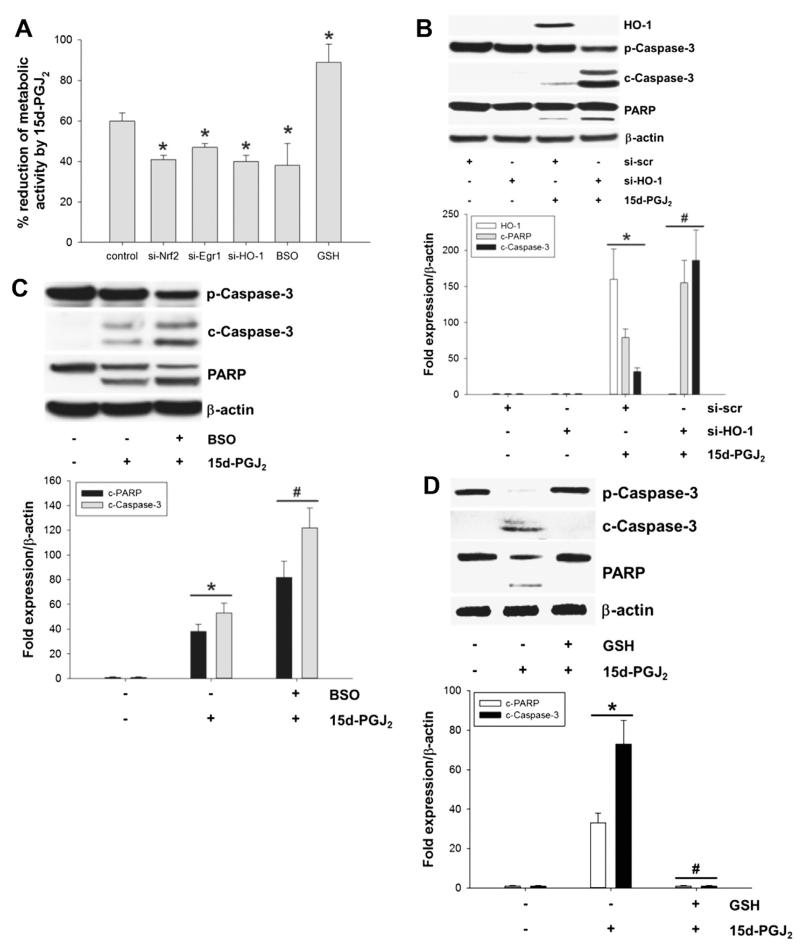
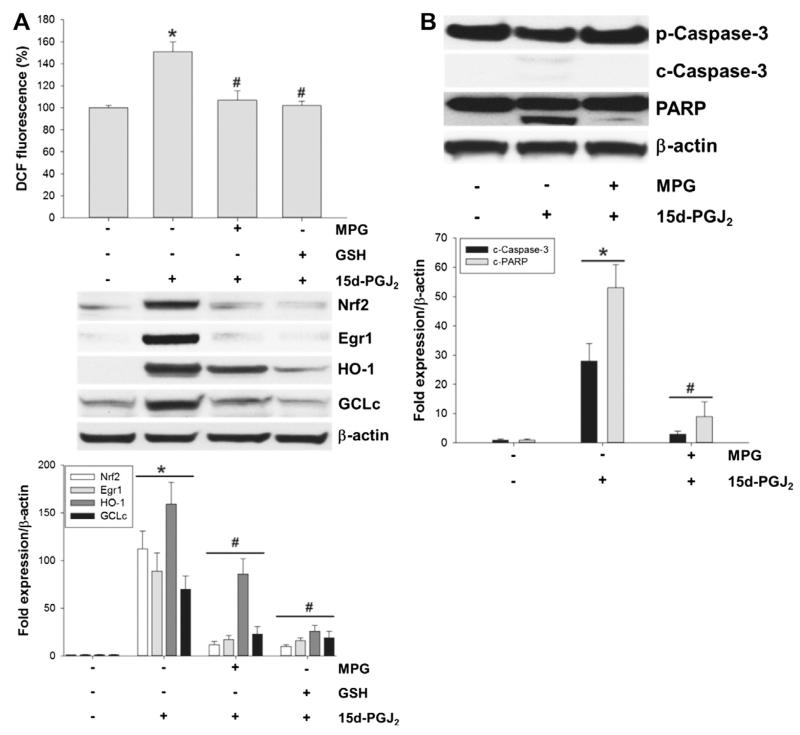
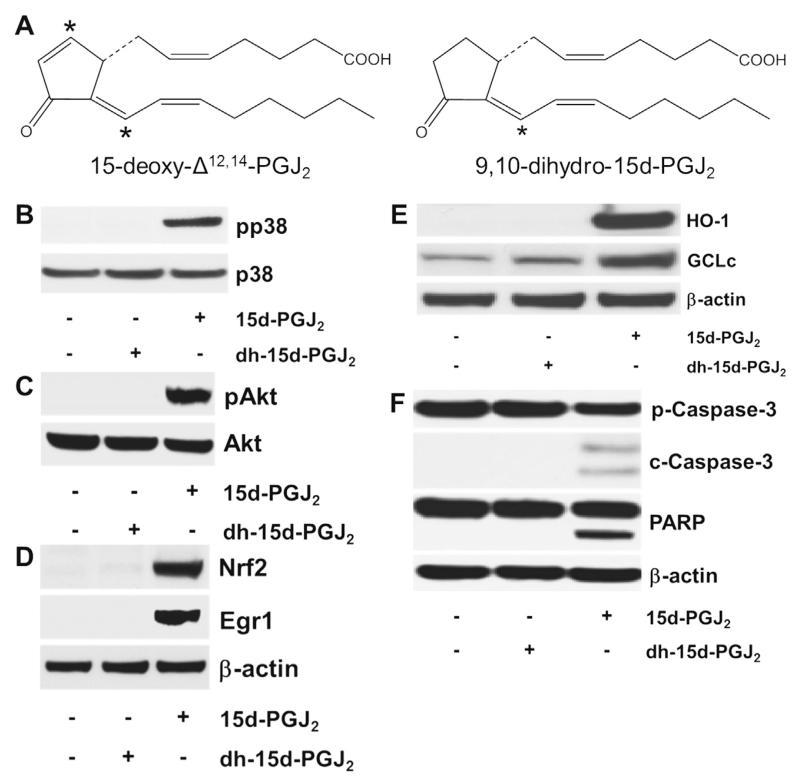
Despite considerable efforts to improve treatment modalities for osteosarcoma (OS), patient survival remains poor mainly due to pro-survival pathways in OS cells. Among others, prostaglandins (PGs) are the potent regulators of bone homoeostasis and OS pathophysiology. Therefore, the present study aimed to elucidate the impact of 15-deoxy-Δ(12,14)-PGJ2 (15d-PGJ2, a stable PGD2 degradation product) on cell death/cell survival pathways in p53-deficient MG-63 OS cells. Our findings show that 15d-PGJ2 induces generation of reactive oxygen species that promote p38 MAPK activation and subsequent Akt phosphorylation. This pathway induced nuclear expression of Nrf2 and Egr1, and increased transcription of haem oxygenase-1 (HO-1) and the catalytic subunit of glutamate cysteine ligase (GCLc), catalysing the first step in GSH synthesis. Silencing of Nrf2, Egr1 and HO-1 significantly elevated 15d-PGJ2-mediated reduction of cellular metabolic activity. Activation of cell survival genes including HO-1 and GCLc inhibited 15d-PGJ2-induced cleavage of pro-caspase-3 and PARP. Annexin V/propidium iodide staining showed an increase in early/late apoptotic cells in response to 15d-PGJ2. The observed 15d-PGJ2-mediated signalling events are independent of PGD2 receptors (DP1 and DP2) and PPARγ. In addition, the electrophilic carbon atom C9 is a prerequisite for the observed activity of 15d-PGJ2. The present data show that the intracellular redox imbalance acted as a node and triggered both death and survival pathways in response to 15d-PGJ2. Pharmacological or genetic interference of the pro-survival pathway, the p38 MAPK/Akt/Nrf2-Egr1/HO-1-GCLc axis, sensitizes MG-63 cells towards 15d-PGJ2-mediated apoptosis.
尽管在改善骨肉瘤(OS)治疗方式方面付出了巨大努力,但患者生存率仍然很低,主要原因是OS细胞中的促生存途径。其中,前列腺素(PGs)是骨稳态和OS病理生理学的有效调节因子。因此,本研究旨在阐明15-脱氧-Δ(12,14)-前列腺素J2(15d-PGJ2,一种稳定的PGD2降解产物)对p53缺陷型MG-63 OS细胞中细胞死亡/细胞生存途径的影响。我们的研究结果表明,15d-PGJ2诱导活性氧的产生,促进p38丝裂原活化蛋白激酶(MAPK)的激活以及随后的Akt磷酸化。该途径诱导核因子E2相关因子2(Nrf2)和早期生长反应蛋白1(Egr1)的核表达,并增加血红素加氧酶-1(HO-1)和谷氨酸半胱氨酸连接酶催化亚基(GCLc)的转录,催化谷胱甘肽(GSH)合成的第一步。Nrf2、Egr1和HO-1的沉默显著提高了15d-PGJ2介导的细胞代谢活性降低。包括HO-1和GCLc在内的细胞生存基因的激活抑制了15d-PGJ2诱导的前半胱天冬酶-3和聚(ADP-核糖)聚合酶(PARP)的裂解。膜联蛋白V/碘化丙啶染色显示,响应15d-PGJ2,早期/晚期凋亡细胞增加。观察到的15d-PGJ2介导的信号事件独立于PGD2受体(DP1和DP2)和过氧化物酶体增殖物激活受体γ(PPARγ)。此外,亲电碳原子C9是观察到的15d-PGJ2活性的先决条件。目前的数据表明,细胞内氧化还原失衡作为一个节点,响应15d-PGJ2触发死亡和生存途径。促生存途径p38 MAPK/Akt/Nrf2-Egr1/HO-1-GCLc轴的药理学或遗传学干扰使MG-63细胞对15d-PGJ2介导的凋亡敏感。