He Hao, Cao Shaolong, Niu Tianhua, Zhou Yu, Zhang Lan, Zeng Yong, Zhu Wei, Wang Yu-ping, Deng Hong-wen
Center for Bioinformatics and Genomics, Department of Biostatistics and Bioinformatics, Tulane University School of Public Health and Tropical Medicine, New Orleans, Louisiana, United States of America.
Department of Biomedical Engineering, Tulane University, New Orleans, Louisiana, United States of America.
PLoS One. 2016 Jan 25;11(1):e0147475. doi: 10.1371/journal.pone.0147475. eCollection 2016.
Existing microarray studies of bone mineral density (BMD) have been critical for understanding the pathophysiology of osteoporosis, and have identified a number of candidate genes. However, these studies were limited by their relatively small sample sizes and were usually analyzed individually. Here, we propose a novel network-based meta-analysis approach that combines data across six microarray studies to identify functional modules from human protein-protein interaction (PPI) data, and highlight several differentially expressed genes (DEGs) and a functional module that may play an important role in BMD regulation in women.
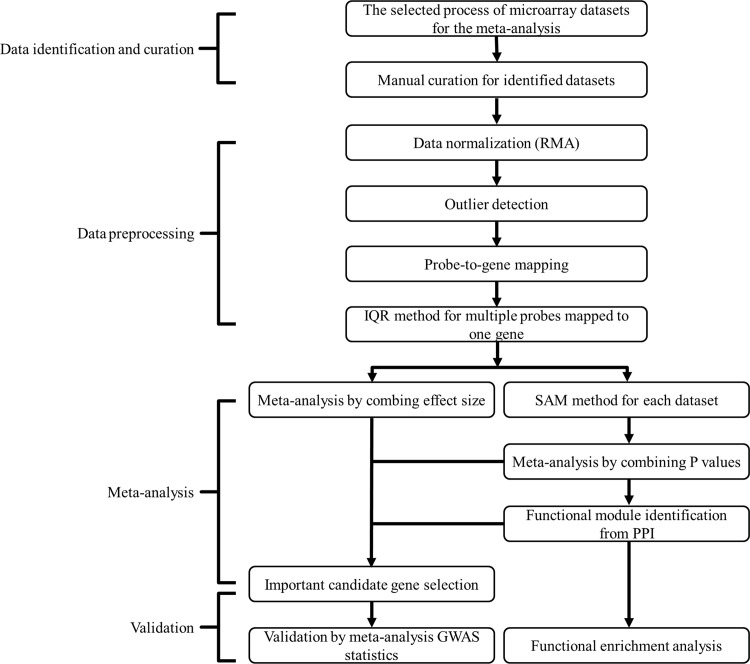
Expression profiling studies were identified by searching PubMed, Gene Expression Omnibus (GEO) and ArrayExpress. Two meta-analysis methods were applied across different gene expression profiling studies. The first, a nonparametric Fisher's method, combined p-values from individual experiments to identify genes with large effect sizes. The second method combined effect sizes from individual datasets into a meta-effect size to gain a higher precision of effect size estimation across all datasets. Genes with Q test's p-values < 0.05 or I(2) values > 50% were assessed by a random effects model and the remainder by a fixed effects model. Using Fisher's combined p-values, functional modules were identified through an integrated analysis of microarray data in the context of large protein-protein interaction (PPI) networks. Two previously published meta-analysis studies of genome-wide association (GWA) datasets were used to determine whether these module genes were genetically associated with BMD. Pathway enrichment analysis was performed with a hypergeometric test.
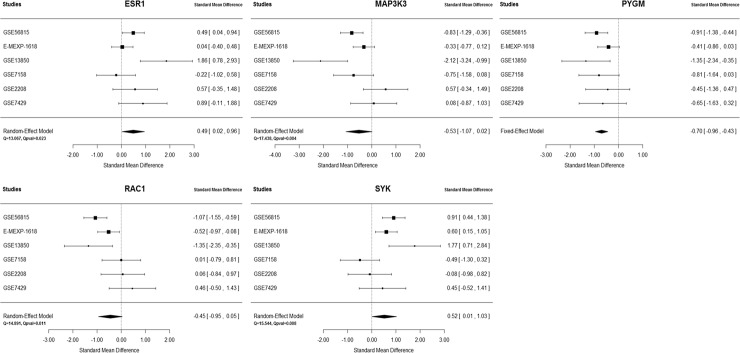
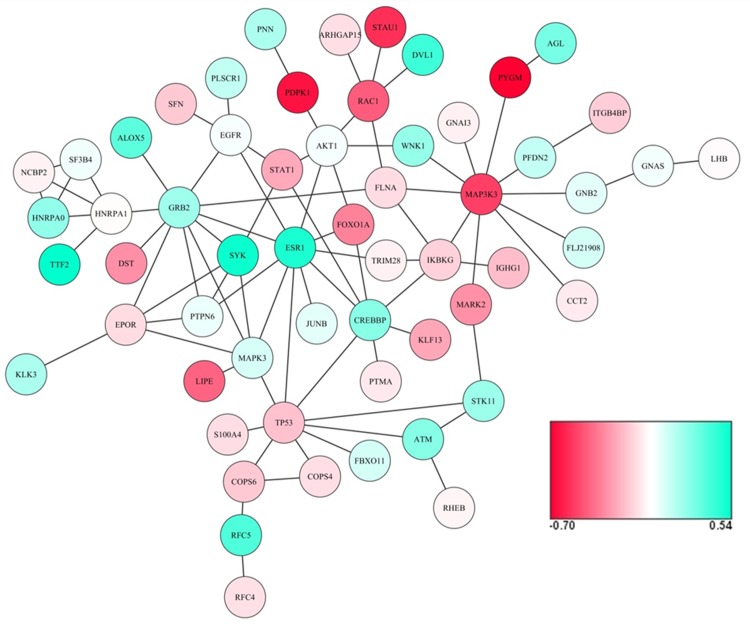
Six gene expression datasets were identified, which included a total of 249 (129 high BMD and 120 low BMD) female subjects. Using a network-based meta-analysis, a consensus module containing 58 genes (nodes) and 83 edges was detected. Pathway enrichment analysis of the 58 module genes revealed that these genes were enriched in several important KEGG pathways including Osteoclast differentiation, B cell receptor signaling pathway, MAPK signaling pathway, Chemokine signaling pathway and Insulin signaling pathway. The importance of module genes was replicated by demonstrating that most module genes were genetically associated with BMD in the GWAS data sets. Meta-analyses were performed at the individual gene level by combining p-values and effect sizes. Five candidate genes (ESR1, MAP3K3, PYGM, RAC1 and SYK) were identified based on gene expression meta-analysis, and their associations with BMD were also replicated by two BMD meta-analysis studies.
In summary, our network-based meta-analysis not only identified important differentially expressed genes but also discovered biologically meaningful functional modules for BMD determination. Our study may provide novel therapeutic targets for osteoporosis in women.
现有的骨密度(BMD)微阵列研究对于理解骨质疏松症的病理生理学至关重要,并已鉴定出许多候选基因。然而,这些研究受到样本量相对较小的限制,并且通常是单独分析的。在此,我们提出一种基于网络的新型荟萃分析方法,该方法整合了六项微阵列研究的数据,以从人类蛋白质 - 蛋白质相互作用(PPI)数据中识别功能模块,并突出显示了几个差异表达基因(DEGs)以及一个可能在女性骨密度调节中起重要作用的功能模块。
通过搜索PubMed、基因表达综合数据库(GEO)和ArrayExpress来识别表达谱研究。在不同的基因表达谱研究中应用了两种荟萃分析方法。第一种是非参数费舍尔方法,它将各个实验的p值合并以识别具有大效应量的基因。第二种方法将各个数据集的效应量合并为一个荟萃效应量,以在所有数据集中获得更高精度的效应量估计。Q检验p值<0.05或I(2)值>50%的基因通过随机效应模型进行评估,其余的通过固定效应模型进行评估。利用费舍尔合并p值,通过在大型蛋白质 - 蛋白质相互作用(PPI)网络背景下对微阵列数据进行综合分析来识别功能模块。使用两项先前发表的全基因组关联(GWA)数据集的荟萃分析研究来确定这些模块基因是否与骨密度存在遗传关联。使用超几何检验进行通路富集分析。
鉴定出六个基因表达数据集,共包括249名女性受试者(129名高骨密度和120名低骨密度)。使用基于网络的荟萃分析,检测到一个包含58个基因(节点)和83条边的共识模块。对这58个模块基因的通路富集分析表明,这些基因在几个重要的KEGG通路中富集,包括破骨细胞分化、B细胞受体信号通路、MAPK信号通路、趋化因子信号通路和胰岛素信号通路。通过证明大多数模块基因在GWAS数据集中与骨密度存在遗传关联,复制了模块基因的重要性。通过合并p值和效应量在个体基因水平上进行荟萃分析。基于基因表达荟萃分析鉴定出五个候选基因(ESR1、MAP3K3、PYGM、RAC1和SYK),并且两项骨密度荟萃分析研究也复制了它们与骨密度的关联。
总之,我们基于网络的荟萃分析不仅鉴定出重要的差异表达基因,还发现了对骨密度测定具有生物学意义的功能模块。我们的研究可能为女性骨质疏松症提供新的治疗靶点。