Lun Aaron T L, Smyth Gordon K
The Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia; Department of Medical Biology, The University of Melbourne, Melbourne, Australia.
The Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia; Department of Mathematics and Statistics, The University of Melbourne, Melbourne, Australia.
F1000Res. 2015 Oct 16;4:1080. doi: 10.12688/f1000research.7016.2. eCollection 2015.
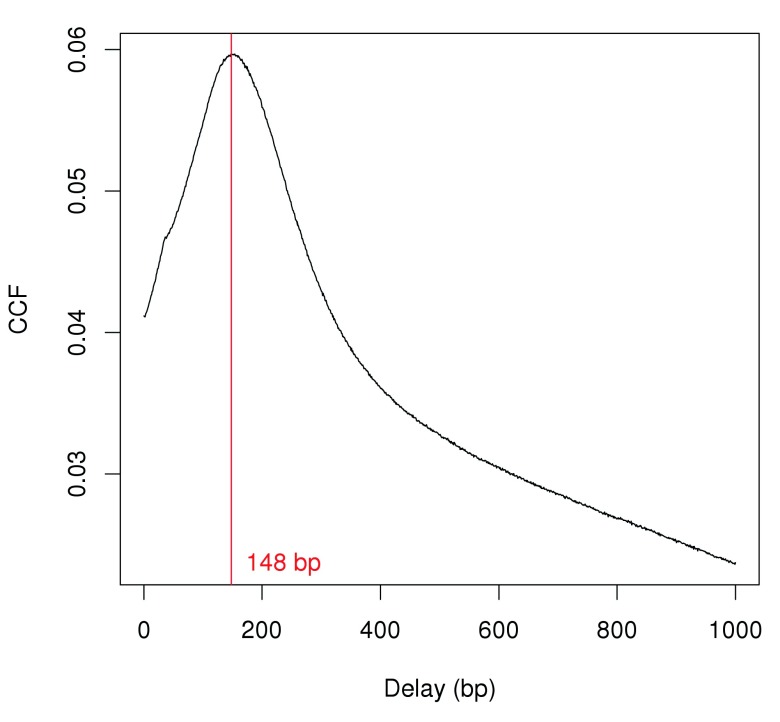
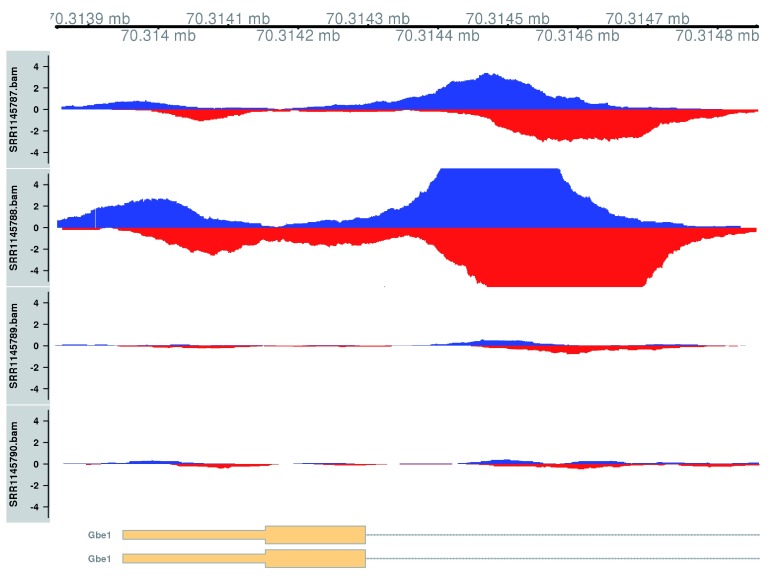
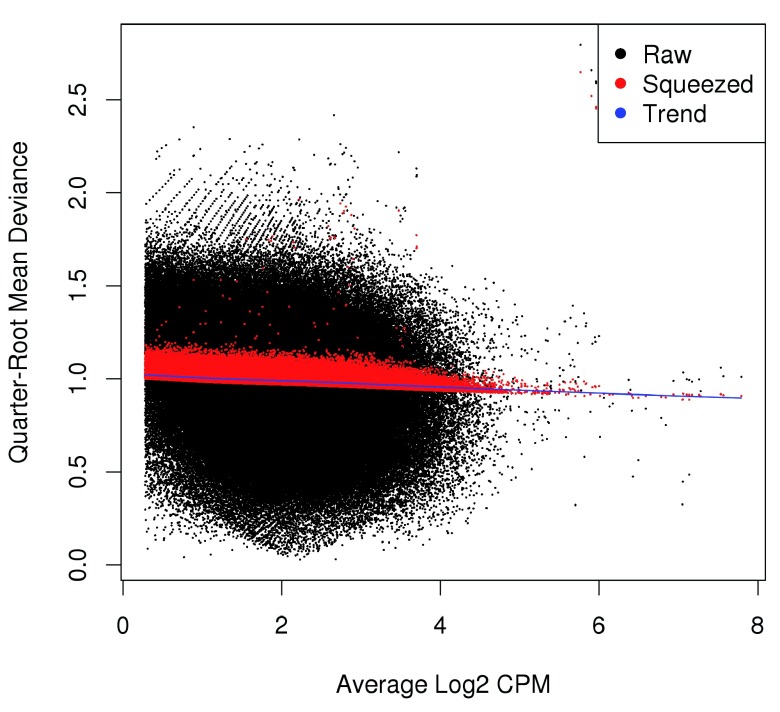
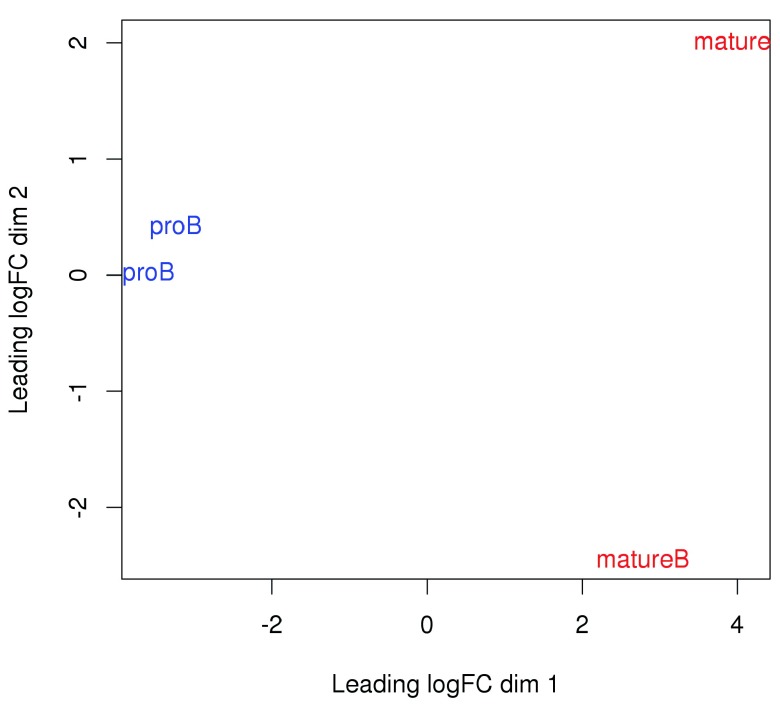
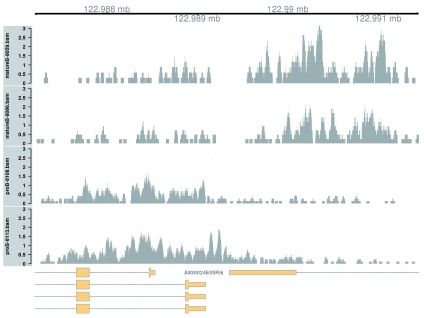
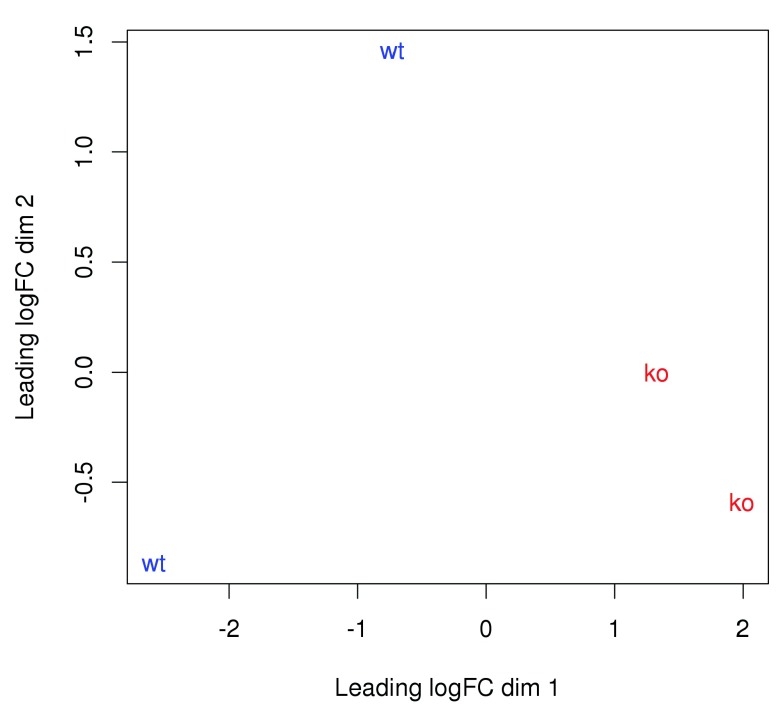
Chromatin immunoprecipitation with massively parallel sequencing (ChIP-seq) is widely used to identify the genomic binding sites for protein of interest. Most conventional approaches to ChIP-seq data analysis involve the detection of the absolute presence (or absence) of a binding site. However, an alternative strategy is to identify changes in the binding intensity between two biological conditions, i.e., differential binding (DB). This may yield more relevant results than conventional analyses, as changes in binding can be associated with the biological difference being investigated. The aim of this article is to facilitate the implementation of DB analyses, by comprehensively describing a computational workflow for the detection of DB regions from ChIP-seq data. The workflow is based primarily on R software packages from the open-source Bioconductor project and covers all steps of the analysis pipeline, from alignment of read sequences to interpretation and visualization of putative DB regions. In particular, detection of DB regions will be conducted using the counts for sliding windows from the csaw package, with statistical modelling performed using methods in the edgeR package. Analyses will be demonstrated on real histone mark and transcription factor data sets. This will provide readers with practical usage examples that can be applied in their own studies.
大规模平行测序染色质免疫沉淀技术(ChIP-seq)被广泛用于识别目标蛋白的基因组结合位点。大多数传统的ChIP-seq数据分析方法涉及检测结合位点的绝对存在(或不存在)。然而,另一种策略是识别两种生物学条件之间结合强度的变化,即差异结合(DB)。与传统分析相比,这可能会产生更相关的结果,因为结合变化可能与所研究的生物学差异相关。本文的目的是通过全面描述从ChIP-seq数据中检测DB区域的计算工作流程,促进DB分析的实施。该工作流程主要基于开源生物导体项目的R软件包,涵盖了分析流程的所有步骤,从读取序列的比对到假定DB区域的解释和可视化。特别是,将使用csaw软件包中滑动窗口的计数来检测DB区域,并使用edgeR软件包中的方法进行统计建模。将在真实的组蛋白标记和转录因子数据集上进行分析。这将为读者提供可应用于他们自己研究的实际使用示例。