Ramo Ana, Quílez Joaquín, Monteagudo Luis, Del Cacho Emilio, Sánchez-Acedo Caridad
Department of Animal Pathology, Faculty of Veterinary Sciences, University of Zaragoza, Zaragoza, Spain.
Department of Anatomy, Embriology and Genetics, Faculty of Veterinary Sciences, University of Zaragoza, Zaragoza, Spain.
PLoS One. 2016 Feb 5;11(2):e0148811. doi: 10.1371/journal.pone.0148811. eCollection 2016.
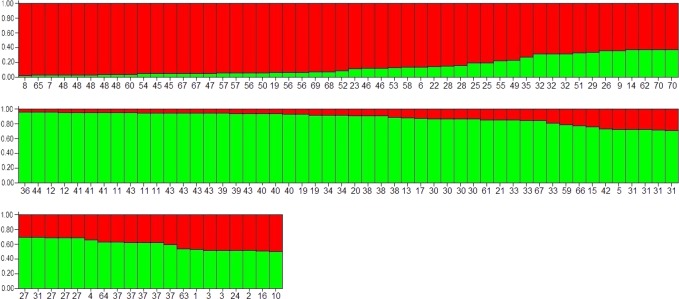
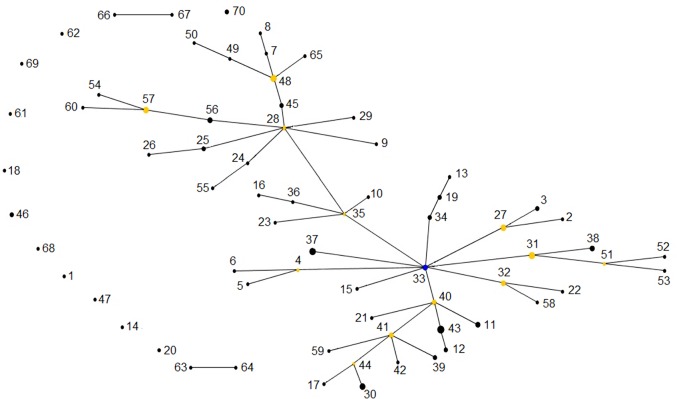
The intra-herd and intra-host genetic variability of 123 Cryptosporidium parvum isolates was investigated using a multilocus fragment typing approach with eleven variable-number tandem-repeat (VNTR) loci and the GP60 gene. Isolates were collected from intensively farmed diarrheic pre-weaned calves originating from 31 dairy farms in three adjoining regions in northern Spain (País Vasco, Cantabria and Asturias). The multilocus tool demonstrated an acceptable typeability, with 104/123 samples amplifying at all twelve loci. The ML2, TP14, GP60 and the previously un-described minisatellite at locus cgd2_3850 were the most discriminatory markers, while others may be dismissed as monomorphic (MSB) or less informative (CP47, ML1 and the novel minisatellites at loci Cgd1_3670 and Cgd6_3940). The 12-satellite typing tool provided a Hunter-Gaston index (HGDI) of 0.987 (95% CI, 0.982-0.992), and differentiated a total of 70 multilocus subtypes (MLTs). The inclusion of only the four most discriminatory markers dramatically reduced the number of MLTs (n: 44) but hardly reduced the HGDI value. A total of 54 MLTs were distinctive for individual farms, indicating that cryptosporidiosis is an endemic condition on most cattle farms. However, a high rate of mixed infections was detected, suggesting frequent meiotic recombination. Namely, multiple MLTs were seen in most farms where several specimens were analyzed (90.5%), with up to 9 MLTs being found on one farm, and individual specimens with mixed populations being reported on 11/29 farms. Bayesian Structure analysis showed that over 35% of isolates had mixed ancestry and analysis of evolutionary descent using the eBURST algorithm detected a high rate (21.4%) of MLTs appearing as singletons, indicating a high degree of genetic divergence. Linkage analysis found evidence of linkage equilibrium and an overall panmictic structure within the C. parvum population in this discrete geographical area.
采用多基因座片段分型方法,利用11个可变数目串联重复(VNTR)基因座和GP60基因,对123株微小隐孢子虫分离株的群体内和宿主体内遗传变异性进行了研究。分离株取自西班牙北部三个毗邻地区(巴斯克地区、坎塔布里亚和阿斯图里亚斯)31个奶牛场的集约化养殖、腹泻的断奶前犊牛。多基因座工具显示出可接受的分型能力,123个样本中有104个在所有12个基因座上均能扩增。ML2、TP14、GP60以及基因座cgd2_3850处先前未描述的小卫星是最具鉴别力的标记,而其他标记可能被视为单态性(MSB)或信息较少(CP47、ML1以及基因座Cgd1_3670和Cgd6_3940处的新型小卫星)。12卫星分型工具的亨特-加斯顿指数(HGDI)为0.987(95%置信区间,0.982 - 0.992),共区分出70种多基因座亚型(MLT)。仅纳入四个最具鉴别力的标记显著减少了MLT的数量(n = 44),但几乎未降低HGDI值。共有54种MLT在各个农场中是独特的,这表明隐孢子虫病在大多数养牛场中呈地方流行状态。然而,检测到较高比例的混合感染,提示频繁的减数分裂重组。具体而言,在大多数分析了多个样本的农场(90.5%)中都发现了多种MLT,在一个农场中最多发现9种MLT,并且在29个农场中的11个农场报告了具有混合群体的个体样本。贝叶斯结构分析表明,超过35%的分离株具有混合谱系,使用eBURST算法对进化谱系进行分析发现,出现单例的MLT比例很高(21.4%),这表明遗传分化程度很高。连锁分析发现了连锁平衡的证据以及该离散地理区域内微小隐孢子虫群体中的总体随机交配结构。