Gelbrich Thomas, Braun Doris E, Griesser Ulrich J
Institute of Pharmacy, University of Innsbruck, Innrain 52c, 6020 Innsbruck, Austria.
Chem Cent J. 2016 Feb 22;10:8. doi: 10.1186/s13065-016-0152-5. eCollection 2016.
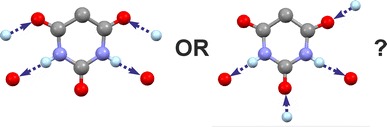
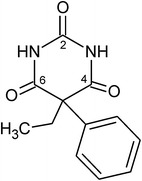
In solid state structures of organic molecules, identical sets of H-bond donor and acceptor functions can result in a range of distinct H-bond connectivity modes. Specifically, competing H-bond structures (HBSs) may differ in the quantitative proportion between one-point and multiple-point H-bond connections. For an assessment of such HBSs, the effects of their internal as well as external (packing) interactions need to be taken into consideration. The semi-classical density sums (SCDS-PIXEL) method, which enables the calculation of interaction energies for molecule-molecule pairs, was used to investigate six polymorphs of phenobarbital (Pbtl) with different quantitative proportions of one-point and two-point H-bond connections.
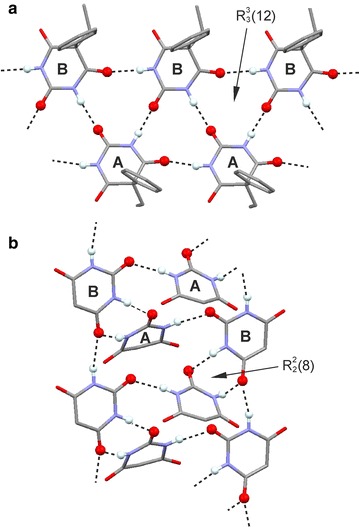
The structures of polymorphs V and VI of Pbtl were determined from single crystal data. Two-point H-bond connections are inherently inflexible in their geometry and lie within a small PIXEL energy range (-45.7 to -49.7 kJ mol(-1)). One-point H-bond connections are geometrically less restricted and subsequently show large variations in their dispersion terms and total energies (-23.1 to -40.5 kJ mol(-1)). The comparison of sums of interaction energies in small clusters containing only the strongest intermolecular interactions showed an advantage for compact HBSs with multiple-point connections, whereas alternative HBSs based on one-point connections may enable more favourable overall packing interactions (i.e. V vs. III). Energy penalties associated with experimental intramolecular geometries relative to the global conformational energy minimum were calculated and used to correct total PIXEL energies. The estimated order of stabilities (based on PIXEL energies) is III > I > II > VI > X > V, with a difference of just 1.7 kJ mol(-1) between the three most stable forms.
For an analysis of competing HBSs, one has to consider the contributions from internal H-bond and non-H-bond interactions, from the packing of multiple HBS instances and intramolecular energy penalties. A compact HBS based on multiple-point H-bond connections should typically lead to more packing alternatives and ultimately to a larger number of viable low-energy structures than a competing one-point HBS (i.e. dimer vs. catemer). Coulombic interaction energies associated with typical short intermolecular C-H···O contact geometries are small in comparison with dispersion effects associated with the packing complementary molecular shapes.Graphical abstractCompeting H-bond motifs can differ markedly in their energy contributions.
在有机分子的固态结构中,相同的氢键供体和受体功能组可导致一系列不同的氢键连接模式。具体而言,相互竞争的氢键结构(HBSs)在单点和多点氢键连接之间的定量比例可能不同。为了评估此类HBSs,需要考虑其内部以及外部(堆积)相互作用的影响。采用能够计算分子-分子对相互作用能的半经典密度和(SCDS-PIXEL)方法,研究了具有不同单点和两点氢键连接定量比例的六种苯巴比妥(Pbtl)多晶型物。
通过单晶数据确定了Pbtl多晶型物V和VI的结构。两点氢键连接在几何形状上本质上是不灵活的,且处于较小的PIXEL能量范围内(-45.7至-49.7 kJ mol⁻¹)。单点氢键连接在几何上限制较少,因此其色散项和总能量变化较大(-23.1至-40.5 kJ mol⁻¹)。对仅包含最强分子间相互作用的小簇中相互作用能总和的比较表明,具有多点连接的紧密HBSs具有优势,而基于单点连接的替代HBSs可能使整体堆积相互作用更有利(即V与III相比)。计算了相对于全局构象能量最小值的实验分子内几何结构相关的能量罚值,并用于校正总PIXEL能量。估计的稳定性顺序(基于PIXEL能量)为III > I > II > VI > X > V,三种最稳定形式之间的差异仅为~1.7 kJ mol⁻¹。
对于相互竞争的HBSs分析,必须考虑来自内部氢键和非氢键相互作用、多个HBS实例的堆积以及分子内能量罚值的贡献。与相互竞争的单点HBS(即二聚体与链状二聚体)相比,基于多点氢键连接的紧密HBS通常应导致更多的堆积选择,并最终产生更多可行的低能量结构。与堆积互补分子形状相关的色散效应相比,与典型的短分子间C-H···O接触几何结构相关的库仑相互作用能较小。
相互竞争的氢键基序在其能量贡献上可能有显著差异。