Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford, UK.
Adult Intensive Care Unit, John Radcliffe Hospital, Oxford, UK.
Lancet Respir Med. 2016 Apr;4(4):259-71. doi: 10.1016/S2213-2600(16)00046-1. Epub 2016 Feb 23.
Effective targeted therapy for sepsis requires an understanding of the heterogeneity in the individual host response to infection. We investigated this heterogeneity by defining interindividual variation in the transcriptome of patients with sepsis and related this to outcome and genetic diversity.
We assayed peripheral blood leucocyte global gene expression for a prospective discovery cohort of 265 adult patients admitted to UK intensive care units with sepsis due to community-acquired pneumonia and evidence of organ dysfunction. We then validated our findings in a replication cohort consisting of a further 106 patients. We mapped genomic determinants of variation in gene transcription between patients as expression quantitative trait loci (eQTL).
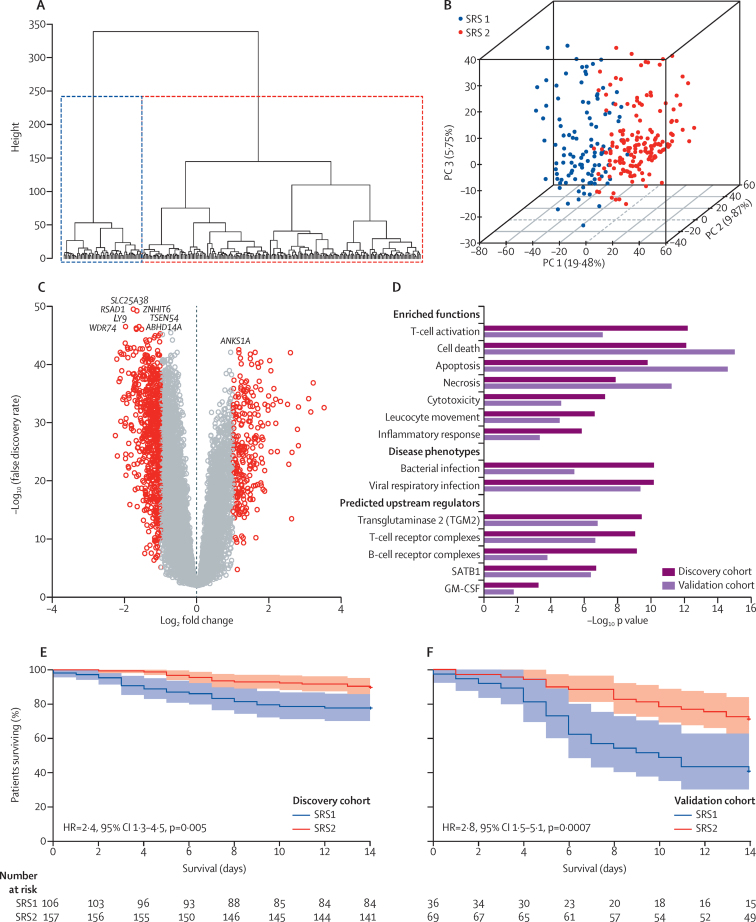
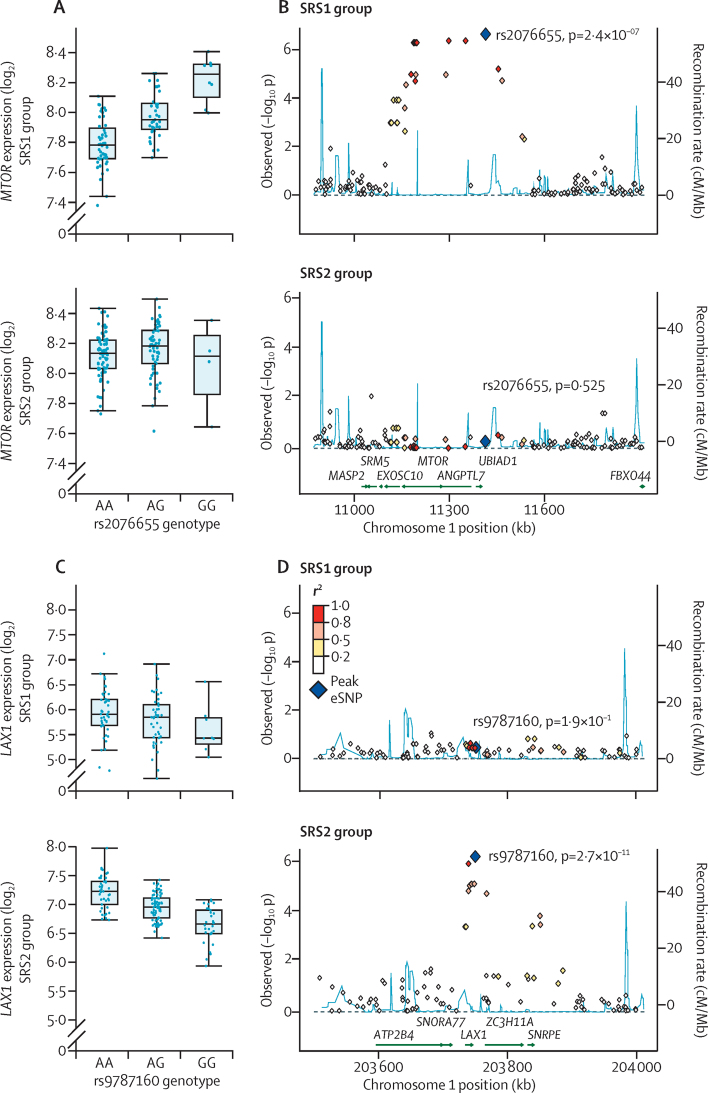
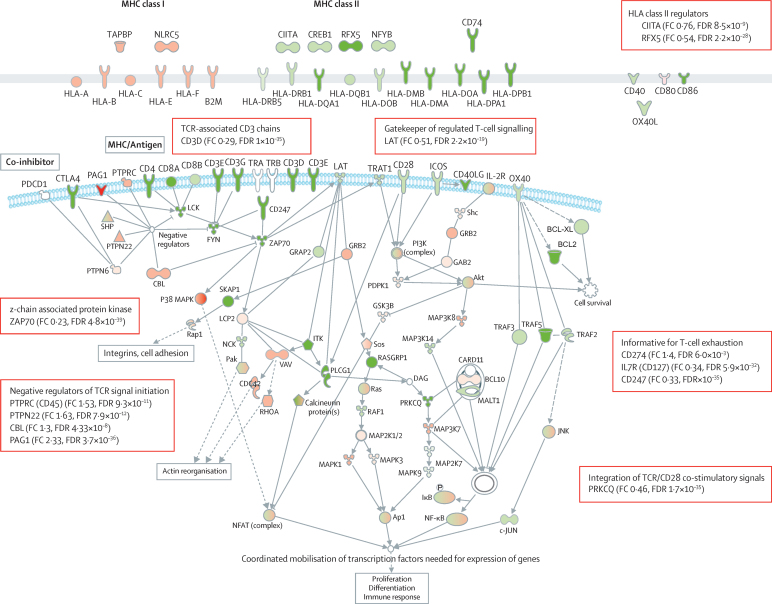
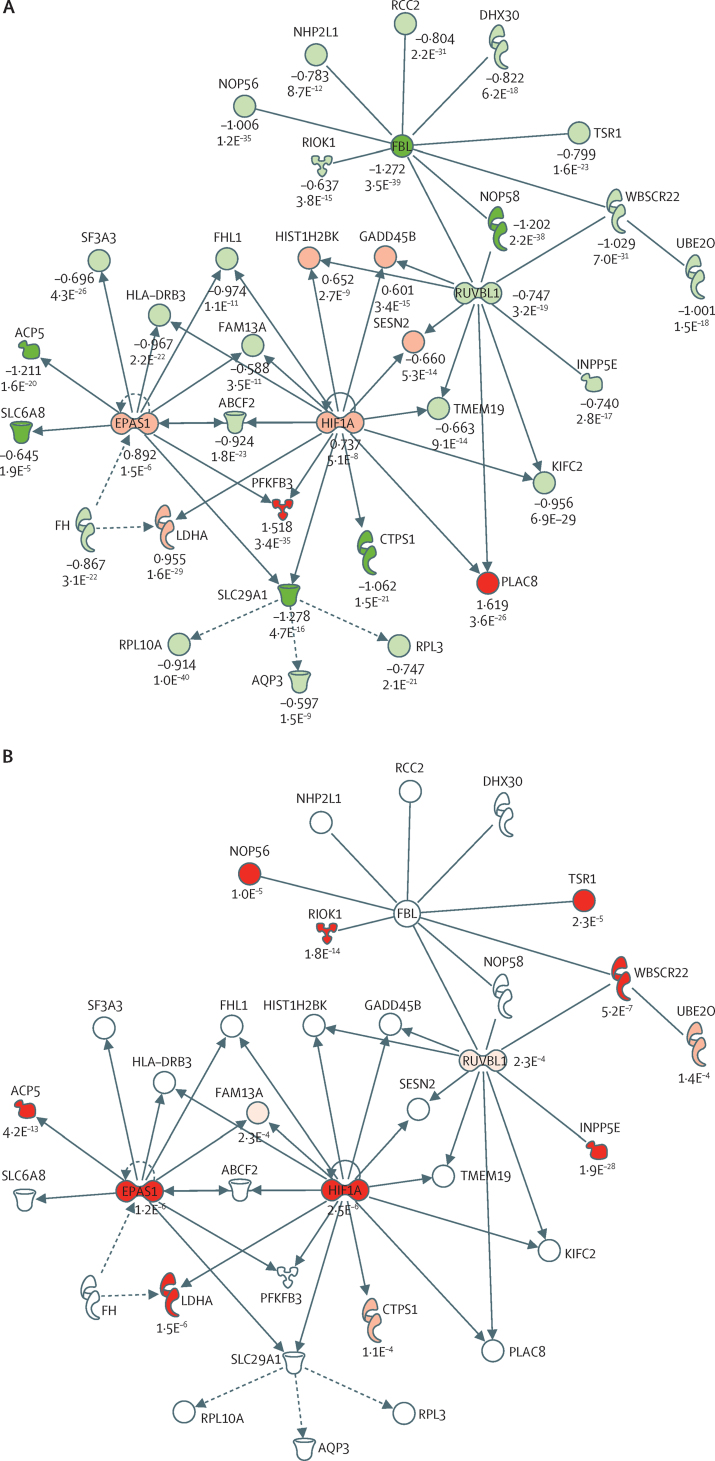
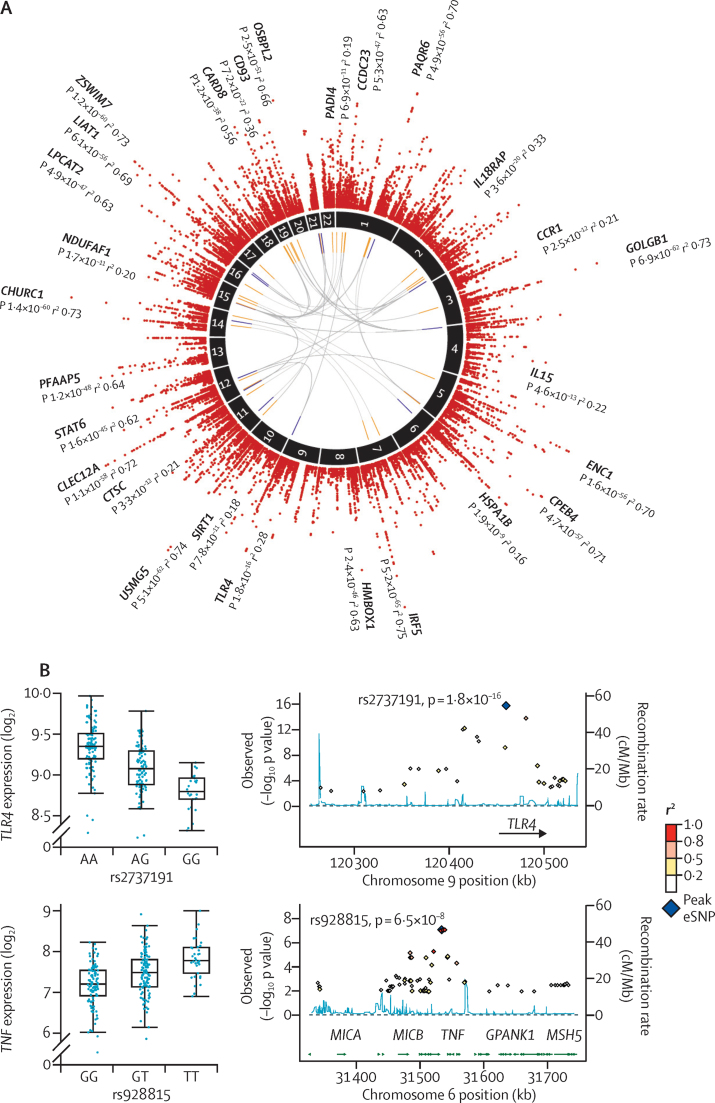
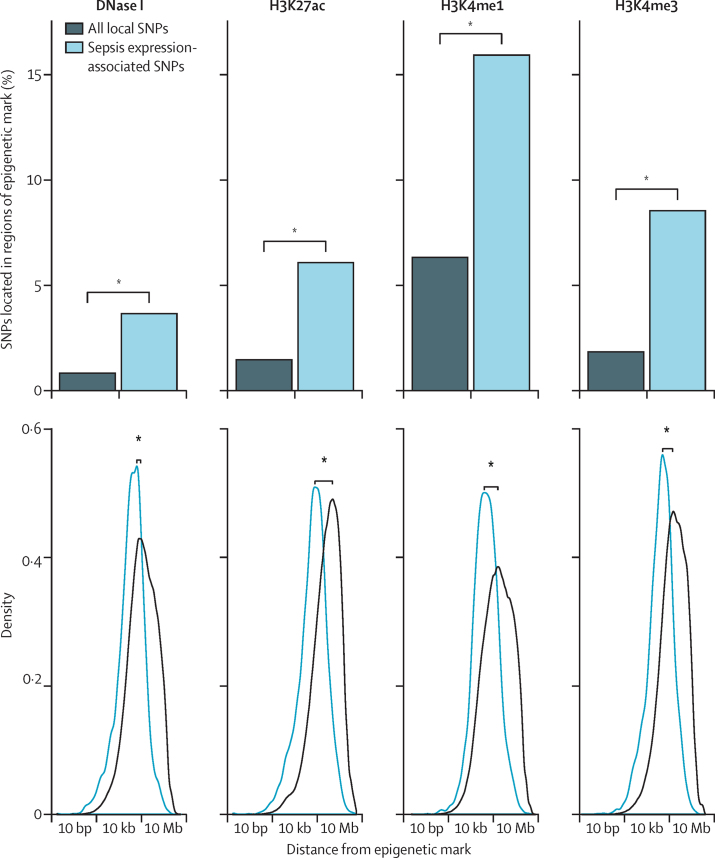
We discovered that following admission to intensive care, transcriptomic analysis of peripheral blood leucocytes defines two distinct sepsis response signatures (SRS1 and SRS2). The presence of SRS1 (detected in 108 [41%] patients in discovery cohort) identifies individuals with an immunosuppressed phenotype that included features of endotoxin tolerance, T-cell exhaustion, and downregulation of human leucocyte antigen (HLA) class II. SRS1 was associated with higher 14 day mortality than was SRS2 (discovery cohort hazard ratio (HR) 2·4, 95% CI 1·3-4·5, p=0·005; validation cohort HR 2·8, 95% CI 1·5-5·1, p=0·0007). We found that a predictive set of seven genes enabled the classification of patients as SRS1 or SRS2. We identified cis-acting and trans-acting eQTL for key immune and metabolic response genes and sepsis response networks. Sepsis eQTL were enriched in endotoxin-induced epigenetic marks and modulated the individual host response to sepsis, including effects specific to SRS group. We identified regulatory genetic variants involving key mediators of gene networks implicated in the hypoxic response and the switch to glycolysis that occurs in sepsis, including HIF1α and mTOR, and mediators of endotoxin tolerance, T-cell activation, and viral defence.
Our integrated genomics approach advances understanding of heterogeneity in sepsis by defining subgroups of patients with different immune response states and prognoses, as well as revealing the role of underlying genetic variation. Our findings provide new insights into the pathogenesis of sepsis and create opportunities for a precision medicine approach to enable targeted therapeutic intervention to improve sepsis outcomes.
European Commission, Medical Research Council (UK), and the Wellcome Trust.
有效的靶向治疗败血症需要了解个体宿主对感染的反应异质性。我们通过定义败血症患者转录组的个体间变异性来研究这种异质性,并将其与预后和遗传多样性相关联。
我们对 265 名因社区获得性肺炎和器官功能障碍而入住英国重症监护病房的成年败血症患者的外周血白细胞进行了全基因组基因表达检测。然后,我们在另外 106 名患者的复制队列中验证了我们的发现。我们将患者之间基因转录变异的基因组决定因素映射为表达数量性状基因座(eQTL)。
我们发现,在入住重症监护病房后,外周血白细胞的转录组分析定义了两种不同的败血症反应特征(SRS1 和 SRS2)。SRS1 的存在(在发现队列中的 108 名[41%]患者中检测到)确定了具有免疫抑制表型的个体,其特征包括内毒素耐受、T 细胞衰竭和人类白细胞抗原(HLA)II 类的下调。SRS1 与 14 天死亡率的相关性高于 SRS2(发现队列危险比(HR)2.4,95%CI 1.3-4.5,p=0.005;验证队列 HR 2.8,95%CI 1.5-5.1,p=0.0007)。我们发现,一组七个基因可以将患者分类为 SRS1 或 SRS2。我们确定了关键免疫和代谢反应基因和败血症反应网络的顺式作用和反式作用 eQTL。败血症 eQTL 在脂多糖诱导的表观遗传标记中富集,并调节个体对败血症的反应,包括对 SRS 组特异的效应。我们鉴定了涉及参与缺氧反应和败血症中糖酵解转换的基因网络的关键介质的调节遗传变异,包括 HIF1α 和 mTOR,以及内毒素耐受、T 细胞激活和病毒防御的介质。
我们的综合基因组学方法通过定义具有不同免疫反应状态和预后的患者亚组来提高对败血症异质性的理解,同时揭示了潜在遗传变异的作用。我们的发现为败血症的发病机制提供了新的见解,并为精准医学方法提供了机会,以实现靶向治疗干预,从而改善败血症的预后。
欧盟委员会、英国医学研究理事会(英国)和惠康信托基金会。