Battistuzzi Fabia U, Schneider Kristan A, Spencer Matthew K, Fisher David, Chaudhry Sophia, Escalante Ananias A
Department of Biological Sciences, Oakland University, Rochester, MI, USA.
Department of MNI, University of Applied Sciences Mittweida, Mittweida, Germany.
BMC Evol Biol. 2016 Feb 29;16:47. doi: 10.1186/s12862-016-0625-0.
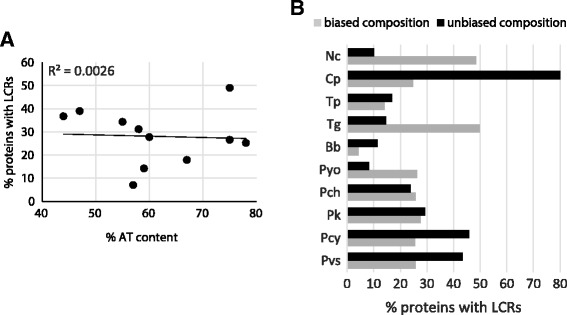
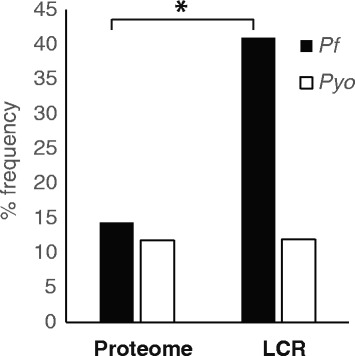
Low complexity regions (LCRs) are a ubiquitous feature in genomes and yet their evolutionary history and functional roles are unclear. Previous studies have shown contrasting evidence in favor of both neutral and selective mechanisms of evolution for different sets of LCRs suggesting that modes of identification of these regions may play a role in our ability to discern their evolutionary history. To further investigate this issue, we used a multiple threshold approach to identify species-specific profiles of proteome complexity and, by comparing properties of these sets, determine the influence that starting parameters have on evolutionary inferences.
We find that, although qualitatively similar, quantitatively each species has a unique LCR profile which represents the frequency of these regions within each genome. Inferences based on these profiles are more accurate in comparative analyses of genome complexity as they allow to determine the relative complexity of multiple genomes as well as the type of repetitiveness that is most common in each. Based on the multiple threshold LCR sets obtained, we identified predominant evolutionary mechanisms at different complexity levels, which show neutral mechanisms acting on highly repetitive LCRs (e.g., homopolymers) and selective forces becoming more important as heterogeneity of the LCRs increases.
Our results show how inferences based on LCRs are influenced by the parameters used to identify these regions. Sets of LCRs are heterogeneous aggregates of regions that include homo- and heteropolymers and, as such, evolve according to different mechanisms. LCR profiles provide a new way to investigate genome complexity across species and to determine the driving mechanism of their evolution.
低复杂度区域(LCRs)是基因组中普遍存在的特征,但其进化历史和功能作用尚不清楚。先前的研究表明,对于不同的LCRs集合,支持中性和选择性进化机制的证据相互矛盾,这表明这些区域的识别模式可能会影响我们辨别其进化历史的能力。为了进一步研究这个问题,我们采用了多阈值方法来识别蛋白质组复杂度的物种特异性图谱,并通过比较这些集合的特性,确定起始参数对进化推断的影响。
我们发现,虽然在质量上相似,但在数量上每个物种都有一个独特的LCR图谱,它代表了每个基因组中这些区域的频率。基于这些图谱的推断在基因组复杂度的比较分析中更准确,因为它们可以确定多个基因组的相对复杂度以及每个基因组中最常见的重复类型。基于获得的多阈值LCR集合,我们确定了不同复杂度水平下的主要进化机制,结果显示中性机制作用于高度重复的LCRs(例如,同聚物),并且随着LCRs异质性增加选择力变得更加重要。
我们的结果表明基于LCRs的推断如何受到用于识别这些区域的参数的影响。LCRs集合是包括同聚物和异聚物在内区域的异质聚集体,因此根据不同机制进化。LCR图谱为研究跨物种基因组复杂度以及确定其进化驱动机制提供了一种新方法。