Bogachev Mikhail I, Kayumov Airat R, Markelov Oleg A, Bunde Armin
Biomedical Engineering Research Center, St. Petersburg Electrotechnical University, St. Petersburg, 197376, Russia.
Department of Genetics, Institute of Fundamental Medicine and Biology, Kazan (Volga Region) Federal University, Kazan, Tatarstan, 420008, Russia.
Sci Rep. 2016 Feb 29;6:22286. doi: 10.1038/srep22286.
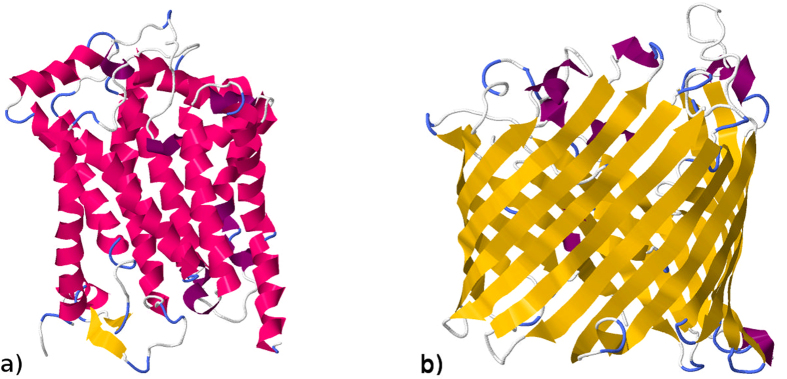
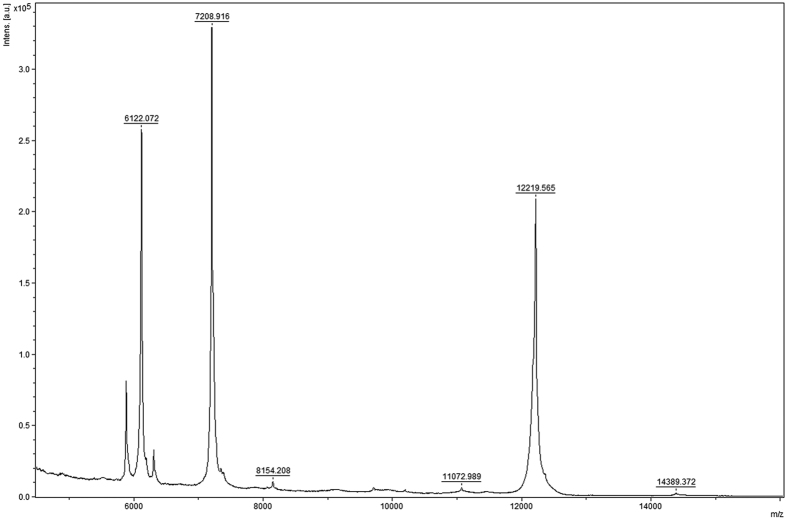
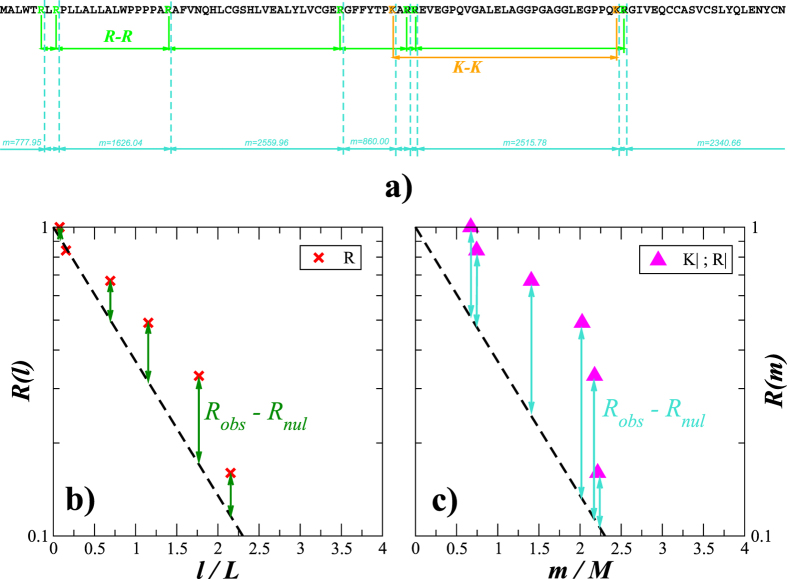
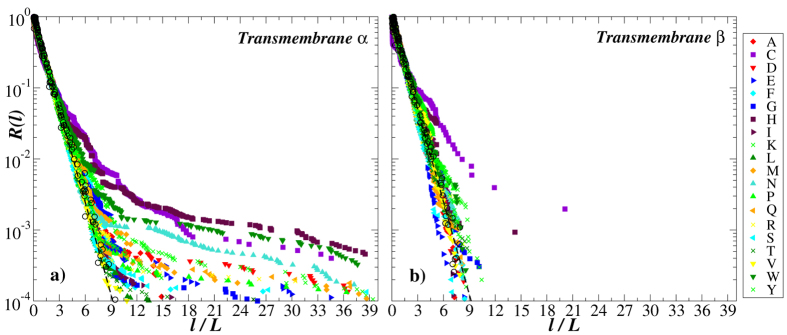
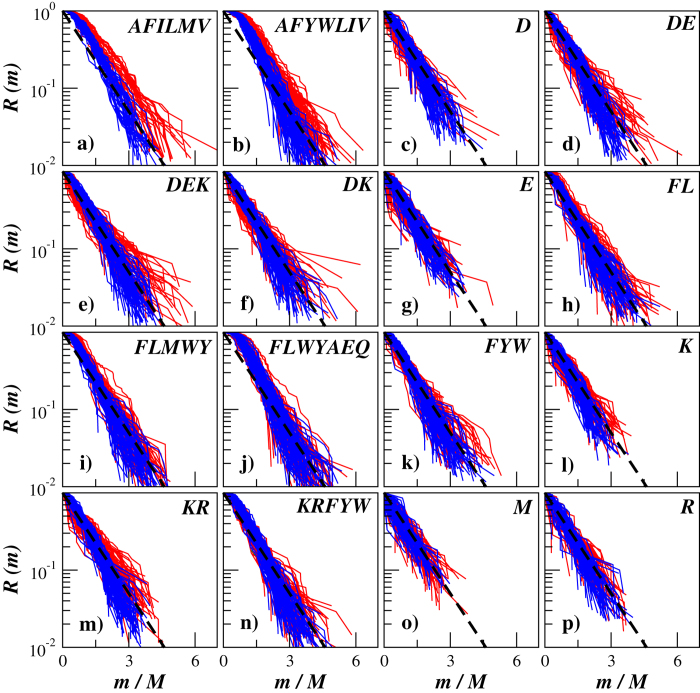
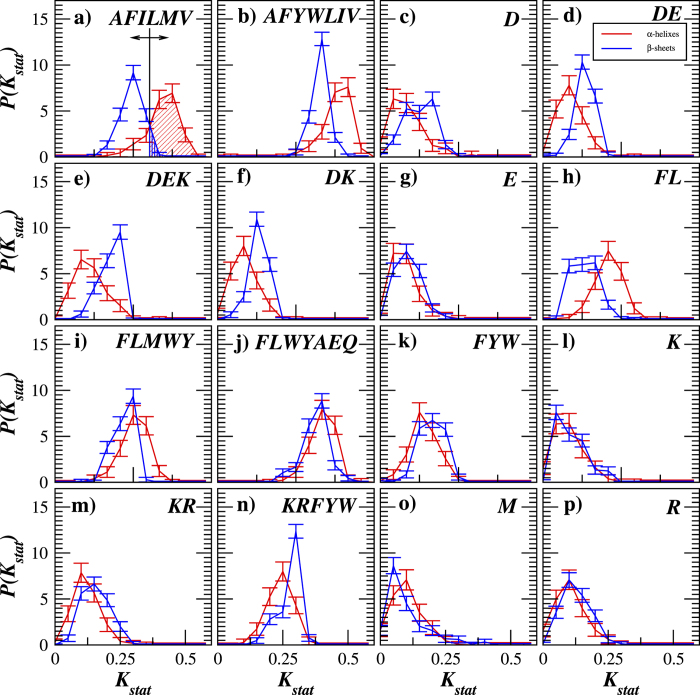
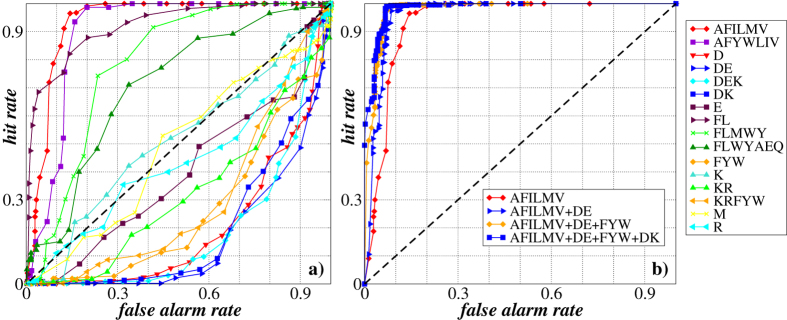
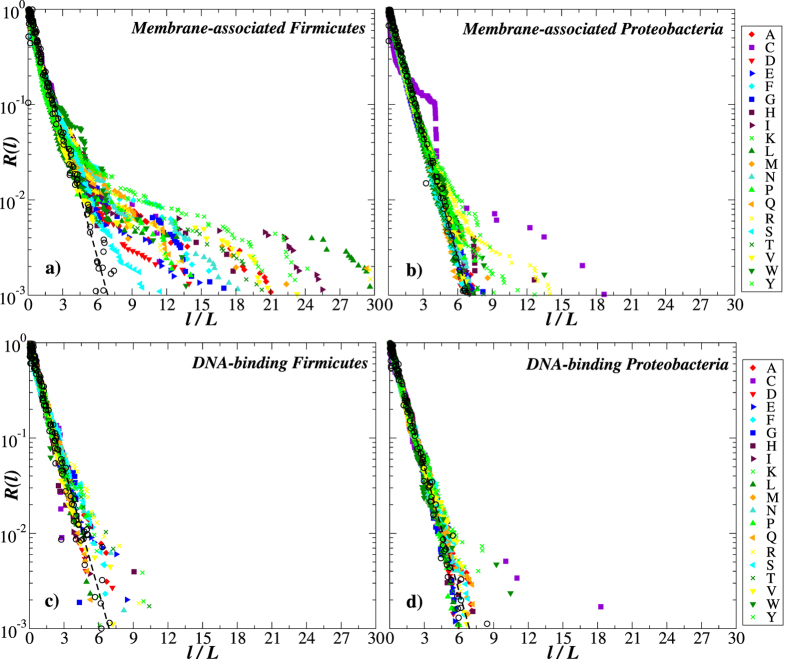
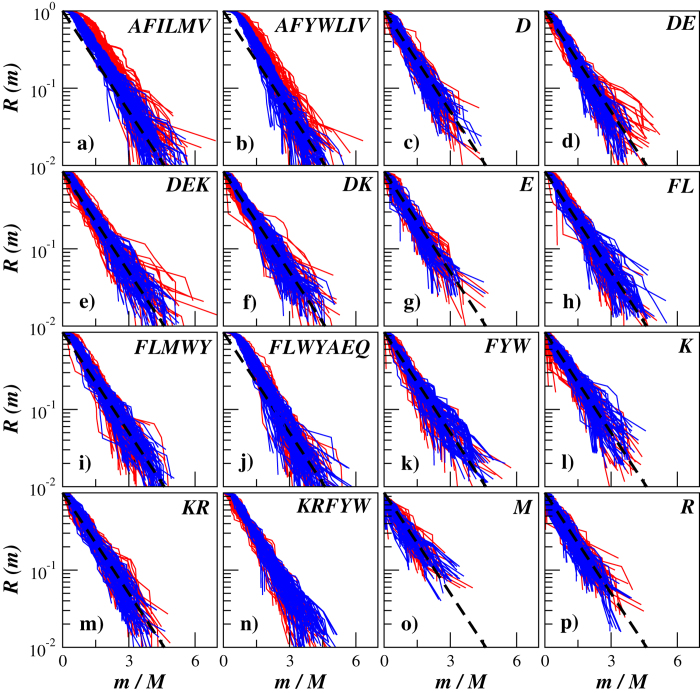
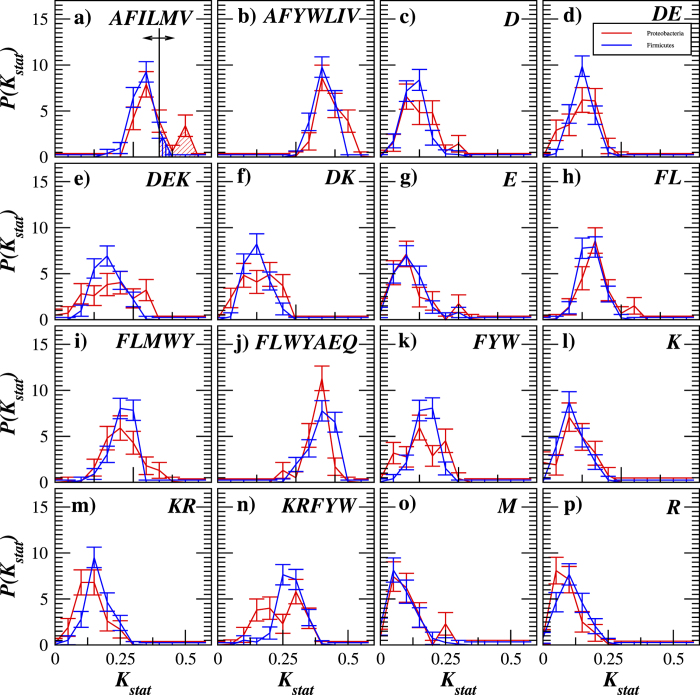
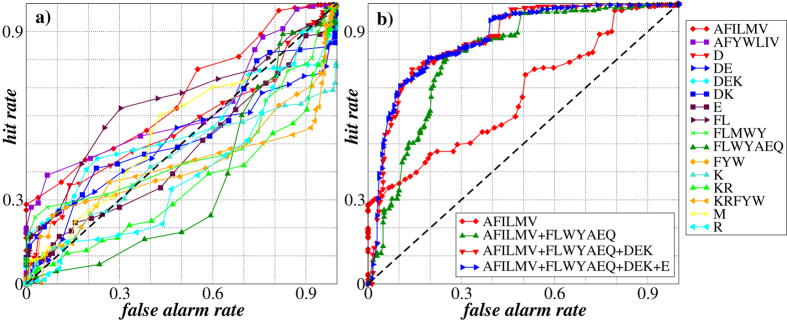
Structural, localization and functional properties of unknown proteins are often being predicted from their primary polypeptide chains using sequence alignment with already characterized proteins and consequent molecular modeling. Here we suggest an approach to predict various structural and structure-associated properties of proteins directly from the mass distributions of their proteolytic cleavage fragments. For amino-acid-specific cleavages, the distributions of fragment masses are determined by the distributions of inter-amino-acid intervals in the protein, that in turn apparently reflect its structural and structure-related features. Large-scale computer simulations revealed that for transmembrane proteins, either α-helical or β -barrel secondary structure could be predicted with about 90% accuracy after thermolysin cleavage. Moreover, 3/4 intrinsically disordered proteins could be correctly distinguished from proteins with fixed three-dimensional structure belonging to all four SCOP structural classes by combining 3-4 different cleavages. Additionally, in some cases the protein cellular localization (cytosolic or membrane-associated) and its host organism (Firmicute or Proteobacteria) could be predicted with around 80% accuracy. In contrast to cytosolic proteins, for membrane-associated proteins exhibiting specific structural conformations, their monotopic or transmembrane localization and functional group (ATP-binding, transporters, sensors and so on) could be also predicted with high accuracy and particular robustness against missing cleavages.
未知蛋白质的结构、定位和功能特性通常是通过与已表征蛋白质的序列比对以及随后的分子建模,从其一级多肽链中预测出来的。在此,我们提出一种直接从蛋白质水解片段的质量分布预测蛋白质各种结构和与结构相关特性的方法。对于氨基酸特异性切割,片段质量的分布由蛋白质中氨基酸间间隔的分布决定,而这反过来显然反映了其结构和与结构相关的特征。大规模计算机模拟表明,对于跨膜蛋白,嗜热菌蛋白酶切割后,α螺旋或β桶二级结构的预测准确率约为90%。此外,通过结合3 - 4种不同的切割方式,四分之三的内在无序蛋白能够与属于所有四个SCOP结构类别的具有固定三维结构的蛋白正确区分开来。此外,在某些情况下,蛋白质的细胞定位(胞质或膜相关)及其宿主生物体(厚壁菌门或变形菌门)的预测准确率可达80%左右。与胞质蛋白不同,对于呈现特定结构构象的膜相关蛋白,其单一位点或跨膜定位以及功能基团(ATP结合、转运蛋白、传感器等)也能够以高精度预测,并且对缺失切割具有特别的稳健性。