Feng Shihai, Lo Chien-Chi, Li Po-E, Chain Patrick S G
Genome Science Group, Bioscience Division, Los Alamos National Laboratory, Los Alamos, NM, 87545, USA.
BMC Bioinformatics. 2016 Feb 29;17:109. doi: 10.1186/s12859-016-0967-z.
Illumina is the most widely used next generation sequencing technology and produces millions of short reads that contain errors. These sequencing errors constitute a major problem in applications such as de novo genome assembly, metagenomics analysis and single nucleotide polymorphism discovery.
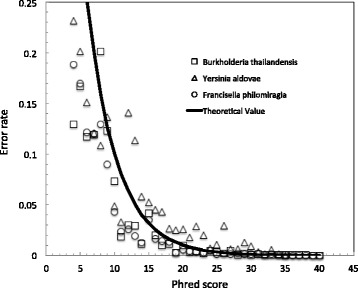
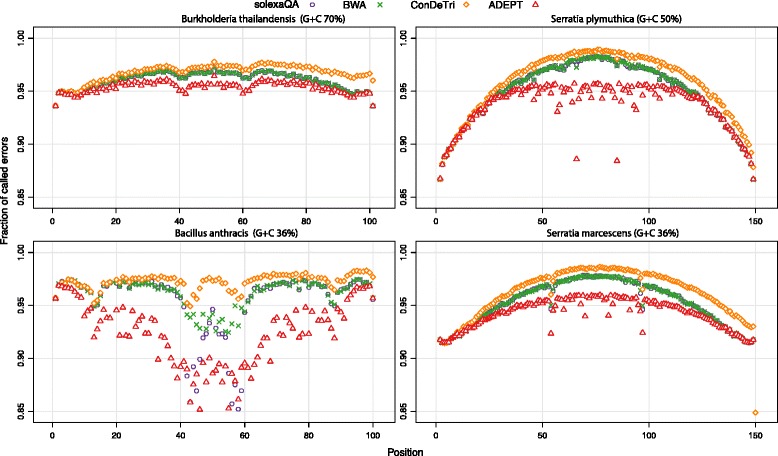
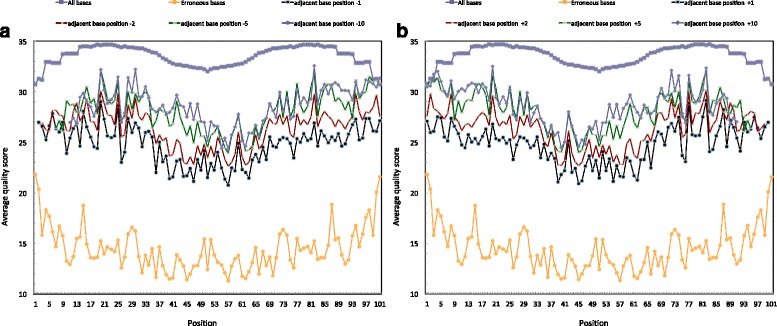
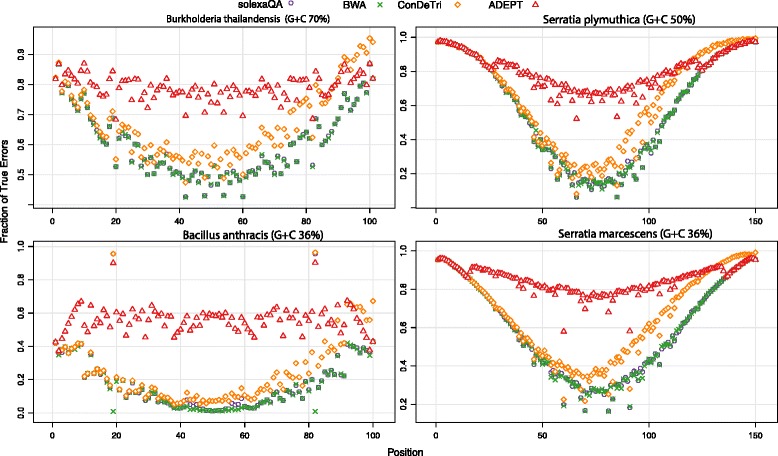
In this study, we present ADEPT, a dynamic error detection method, based on the quality scores of each nucleotide and its neighboring nucleotides, together with their positions within the read and compares this to the position-specific quality score distribution of all bases within the sequencing run. This method greatly improves upon other available methods in terms of the true positive rate of error discovery without affecting the false positive rate, particularly within the middle of reads.
ADEPT is the only tool to date that dynamically assesses errors within reads by comparing position-specific and neighboring base quality scores with the distribution of quality scores for the dataset being analyzed. The result is a method that is less prone to position-dependent under-prediction, which is one of the most prominent issues in error prediction. The outcome is that ADEPT improves upon prior efforts in identifying true errors, primarily within the middle of reads, while reducing the false positive rate.
Illumina是应用最广泛的新一代测序技术,能产生数百万条包含错误的短读段。这些测序错误在诸如从头基因组组装、宏基因组学分析和单核苷酸多态性发现等应用中构成了一个主要问题。
在本研究中,我们提出了ADEPT,一种动态错误检测方法,它基于每个核苷酸及其相邻核苷酸的质量得分,以及它们在读取片段中的位置,并将其与测序运行中所有碱基的位置特异性质量得分分布进行比较。该方法在错误发现的真阳性率方面比其他现有方法有很大改进,同时不影响假阳性率,特别是在读取片段的中间部分。
ADEPT是迄今为止唯一一种通过将位置特异性和相邻碱基质量得分与所分析数据集的质量得分分布进行比较来动态评估读取片段内错误的工具。结果是一种不太容易出现位置依赖性预测不足的方法,而位置依赖性预测不足是错误预测中最突出的问题之一。结果是ADEPT在识别真正错误方面比之前的方法有所改进,主要是在读取片段的中间部分,同时降低了假阳性率。