Karolinska Institutet, Department NVS, Center for Alzheimer Research, Division for Neurogeriatrics, Karolinska Institutet, SE-141 57, Huddinge, Sweden.
Roche Pharma Research and Early Development, NORD DTA, Roche Innovation Center Basel, Basel, Switzerland.
Acta Neuropathol Commun. 2016 Mar 2;4:22. doi: 10.1186/s40478-016-0292-9.
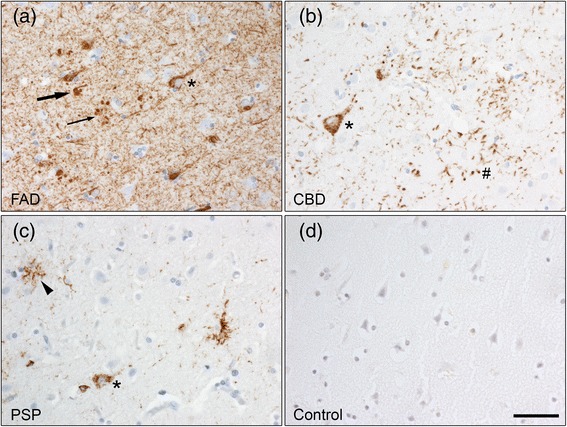
The accumulation of insoluble proteins within neurons and glia cells is a pathological hallmark of several neurodegenerative diseases. Abnormal aggregation of the microtubule-associated protein tau characterizes the neuropathology of tauopathies, such as Alzheimer disease (AD), corticobasal degeneration (CBD) and progressive supranuclear palsy (PSP). An impairment of the lysosomal degradation pathway called macroautophagy, hereafter referred to as autophagy, could contribute to the accumulation of aggregated proteins. The role of autophagy in neurodegeneration has been intensively studied in the context of AD but there are few studies in other tauopathies and it is not known if defects in autophagy is a general feature of tauopathies. In the present study, we analysed autophagic and lysosomal markers in human post-mortem brain samples from patients with early-onset familial AD (FAD) with the APP Swedish mutation (APPswe), CBD and PSP and control individuals.
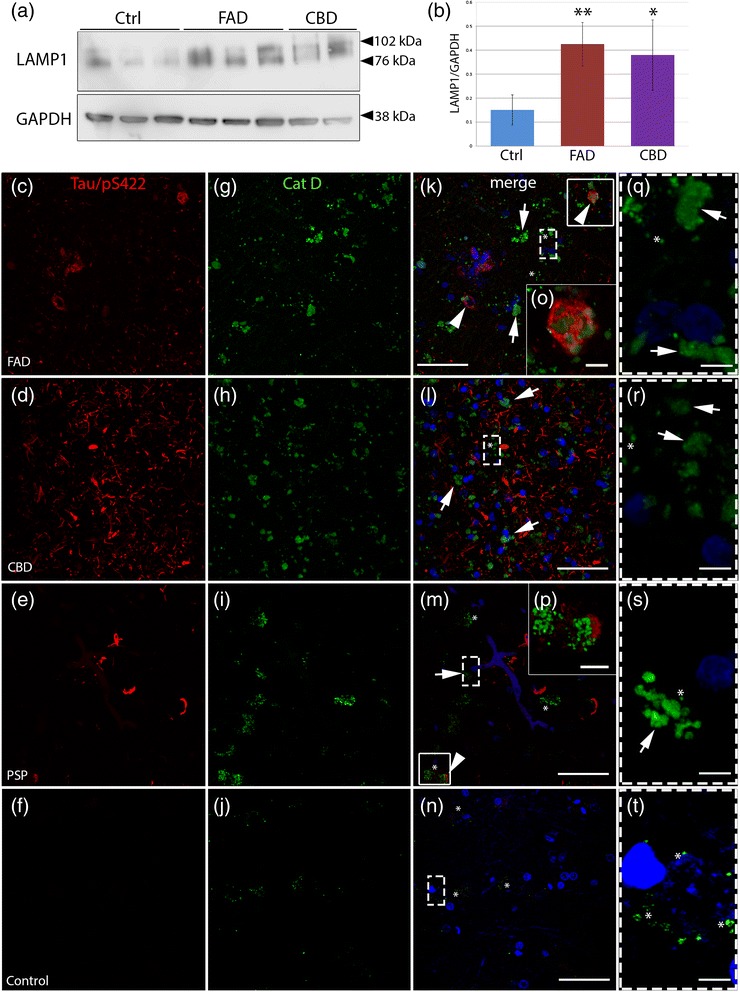
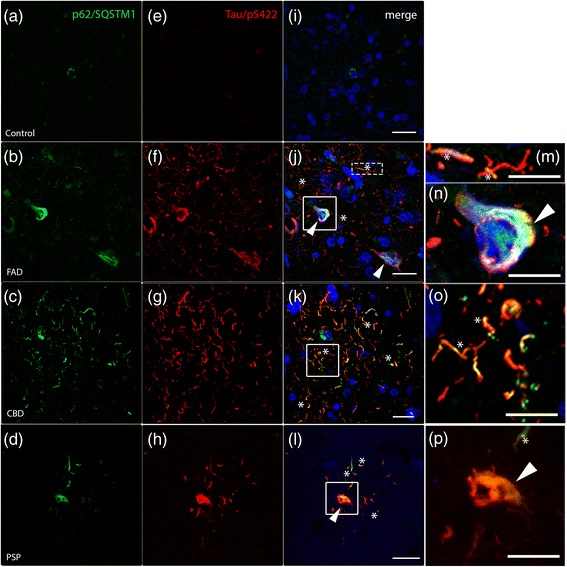
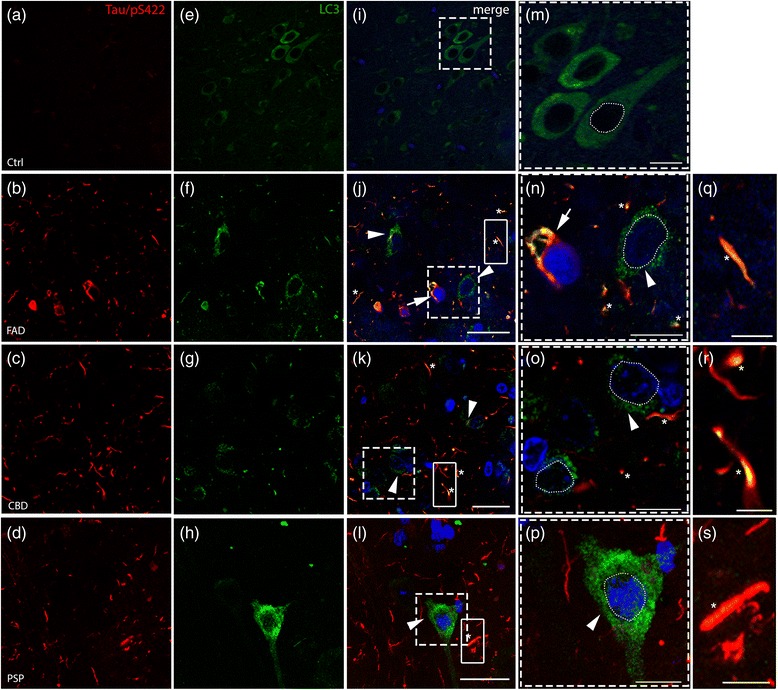
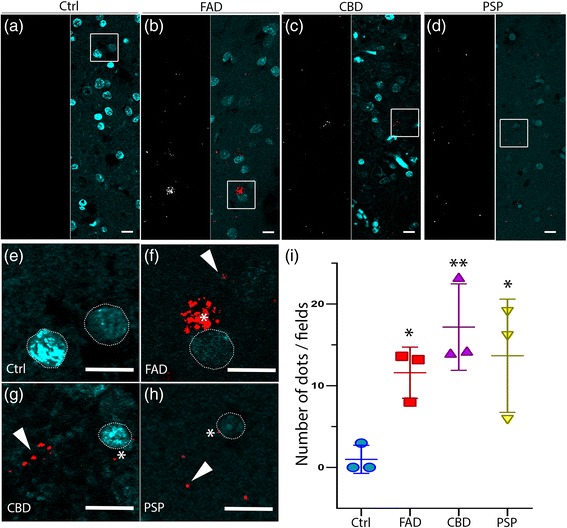
FAD, CBD and PSP patients displayed an increase in LC3-positive vesicles in frontal cortex, indicating an accumulation of autophagic vesicles. Moreover, using double-immunohistochemistry and in situ proximity ligation assay, we observed colocalization of hyperphosphorylated tau with the autophagy marker LC3 in FAD, CBD and PSP patients but not in control individuals. Increased levels of the lysosomal marker LAMP1 was detected in FAD and CBD, and in addition Cathepsin D was diffusely spread in the cytoplasm in all tauopathies suggesting an impaired lysosomal integrity.
Taken together, our results indicate an accumulation of autophagic and lysosomal markers in human brain tissue from patients with primary tauopathies (CBD and PSP) as well as FAD, suggesting a defect of the autophagosome-lysosome pathway that may contribute to the development of tau pathology.
神经元和神经胶质细胞中不溶性蛋白质的积累是几种神经退行性疾病的病理学标志。微管相关蛋白 tau 的异常聚集特征是 tau 病的神经病理学,如阿尔茨海默病 (AD)、皮质基底节变性 (CBD) 和进行性核上性麻痹 (PSP)。溶酶体降解途径的损伤称为巨自噬,以下简称自噬,可能导致聚集蛋白的积累。自噬在神经退行性变中的作用在 AD 背景下得到了深入研究,但在其他 tau 病中研究较少,也不知道自噬缺陷是否是 tau 病的普遍特征。在本研究中,我们分析了早发性家族性 AD (FAD) 患者、CBD 和 PSP 患者以及对照个体的人死后大脑样本中的自噬和溶酶体标志物。
FAD、CBD 和 PSP 患者的额皮质中 LC3 阳性囊泡增加,表明自噬囊泡的积累。此外,通过双重免疫组织化学和原位接近连接测定,我们观察到在 FAD、CBD 和 PSP 患者中,过度磷酸化的 tau 与自噬标记物 LC3 共定位,但在对照个体中未观察到。在 FAD 和 CBD 中检测到溶酶体标记物 LAMP1 的水平增加,此外,在所有 tau 病中,组织蛋白酶 D 弥漫性分布在细胞质中,表明溶酶体完整性受损。
总之,我们的结果表明,原发性 tau 病 (CBD 和 PSP) 以及 FAD 患者的人脑组织中积累了自噬和溶酶体标志物,表明自噬体-溶酶体途径的缺陷可能导致 tau 病理学的发展。