Pérez-Rodríguez Javier, García-Pedrajas Nicolás
Department of Computing and Numerical Analysis, University of Córdoba, Córdoba, 14071, Campus de Rabanales, Spain.
BMC Bioinformatics. 2016 Mar 5;17:117. doi: 10.1186/s12859-016-0968-y.
Recognizing the different functional parts of genes, such as promoters, translation initiation sites, donors, acceptors and stop codons, is a fundamental task of many current studies in Bioinformatics. Currently, the most successful methods use powerful classifiers, such as support vector machines with various string kernels. However, with the rapid evolution of our ability to collect genomic information, it has been shown that combining many sources of evidence is fundamental to the success of any recognition task. With the advent of next-generation sequencing, the number of available genomes is increasing very rapidly. Thus, methods for making use of such large amounts of information are needed.
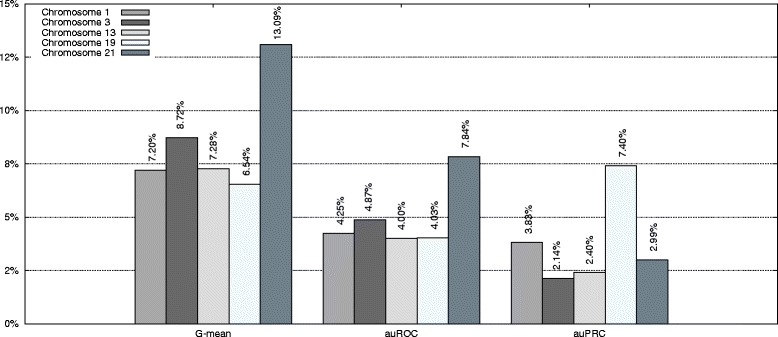
In this paper, we present a methodology for combining tens or even hundreds of different classifiers for an improved performance. Our approach can include almost a limitless number of sources of evidence. We can use the evidence for the prediction of sites in a certain species, such as human, or other species as needed. This approach can be used for any of the functional recognition tasks cited above. However, to provide the necessary focus, we have tested our approach in two functional recognition tasks: translation initiation site and stop codon recognition. We have used the entire human genome as a target and another 20 species as sources of evidence and tested our method on five different human chromosomes. The proposed method achieves better accuracy than the best state-of-the-art method both in terms of the geometric mean of the specificity and sensitivity and the area under the receiver operating characteristic and precision recall curves. Furthermore, our approach shows a more principled way for selecting the best genomes to be combined for a given recognition task.
Our approach has proven to be a powerful tool for improving the performance of functional site recognition, and it is a useful method for combining many sources of evidence for any recognition task in Bioinformatics. The results also show that the common approach of heuristically choosing the species to be used as source of evidence can be improved because the best combinations of genomes for recognition were those not usually selected. Although the experiments were performed for translation initiation site and stop codon recognition, any other recognition task may benefit from our methodology.
识别基因的不同功能部分,如启动子、翻译起始位点、供体、受体和终止密码子,是当前许多生物信息学研究的一项基本任务。目前,最成功的方法使用强大的分类器,如带有各种字符串核的支持向量机。然而,随着我们收集基因组信息能力的快速发展,已经表明结合多种证据来源是任何识别任务成功的基础。随着下一代测序技术的出现,可用基因组的数量正在迅速增加。因此,需要利用如此大量信息的方法。
在本文中,我们提出了一种结合数十个甚至数百个不同分类器以提高性能的方法。我们的方法几乎可以包含无限数量的证据来源。我们可以根据需要使用某个物种(如人类)或其他物种中位点预测的证据。这种方法可用于上述任何功能识别任务。然而,为了提供必要的重点,我们在两个功能识别任务中测试了我们的方法:翻译起始位点和终止密码子识别。我们以整个人类基因组为目标,以另外20个物种作为证据来源,并在五条不同的人类染色体上测试了我们的方法。所提出的方法在特异性和敏感性的几何平均值以及接收器操作特征曲线和精确召回曲线下的面积方面都比最佳的现有方法具有更高的准确性。此外,我们的方法为选择用于给定识别任务的最佳组合基因组提供了一种更有原则的方式。
我们的方法已被证明是提高功能位点识别性能的强大工具,并且是一种用于结合生物信息学中任何识别任务的多种证据来源的有用方法。结果还表明,启发式选择用作证据来源的物种的常见方法可以得到改进,因为用于识别的基因组的最佳组合是那些通常未被选择的组合。尽管实验是针对翻译起始位点和终止密码子识别进行的,但任何其他识别任务都可能从我们的方法中受益。