Sonnenburg Sören, Schweikert Gabriele, Philips Petra, Behr Jonas, Rätsch Gunnar
Fraunhofer Institute FIRST, Kekuléstr, 7, 12489 Berlin, Germany.
BMC Bioinformatics. 2007;8 Suppl 10(Suppl 10):S7. doi: 10.1186/1471-2105-8-S10-S7.
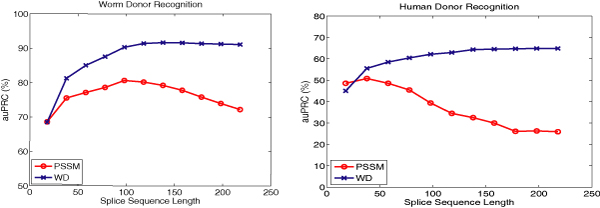
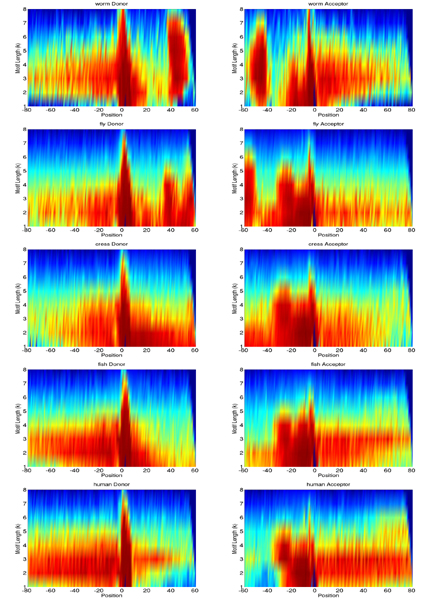
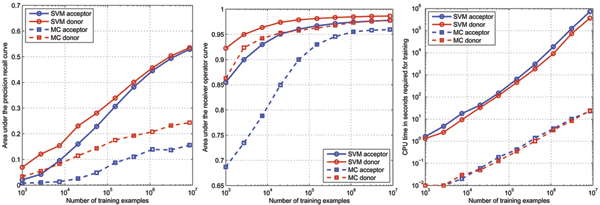
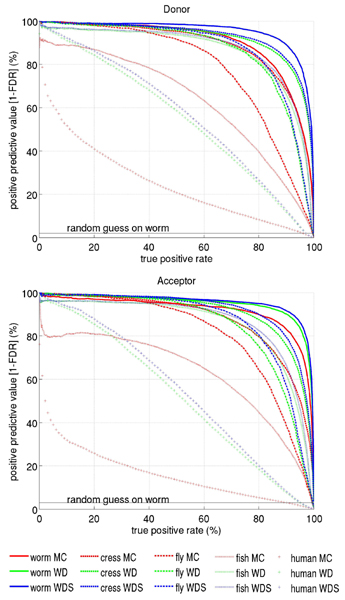
For splice site recognition, one has to solve two classification problems: discriminating true from decoy splice sites for both acceptor and donor sites. Gene finding systems typically rely on Markov Chains to solve these tasks.
In this work we consider Support Vector Machines for splice site recognition. We employ the so-called weighted degree kernel which turns out well suited for this task, as we will illustrate in several experiments where we compare its prediction accuracy with that of recently proposed systems. We apply our method to the genome-wide recognition of splice sites in Caenorhabditis elegans, Drosophila melanogaster, Arabidopsis thaliana, Danio rerio, and Homo sapiens. Our performance estimates indicate that splice sites can be recognized very accurately in these genomes and that our method outperforms many other methods including Markov Chains, GeneSplicer and SpliceMachine. We provide genome-wide predictions of splice sites and a stand-alone prediction tool ready to be used for incorporation in a gene finder.
Data, splits, additional information on the model selection, the whole genome predictions, as well as the stand-alone prediction tool are available for download at http://www.fml.mpg.de/raetsch/projects/splice.
对于剪接位点识别,必须解决两个分类问题:区分受体和供体位点的真实剪接位点与诱饵剪接位点。基因发现系统通常依靠马尔可夫链来解决这些任务。
在这项工作中,我们考虑使用支持向量机进行剪接位点识别。我们采用了所谓的加权度核,结果证明它非常适合这项任务,正如我们将在几个实验中说明的那样,在这些实验中我们将其预测准确性与最近提出的系统的预测准确性进行了比较。我们将我们的方法应用于秀丽隐杆线虫、黑腹果蝇、拟南芥、斑马鱼和智人的全基因组剪接位点识别。我们的性能评估表明,在这些基因组中可以非常准确地识别剪接位点,并且我们的方法优于许多其他方法,包括马尔可夫链、基因剪接器和剪接机。我们提供了全基因组剪接位点预测以及一个独立的预测工具,可随时用于整合到基因发现器中。
数据、分割、关于模型选择的附加信息、全基因组预测以及独立预测工具可在http://www.fml.mpg.de/raetsch/projects/splice下载。