Christensen Dan Ploug, Ejlerskov Patrick, Rasmussen Izabela, Vilhardt Frederik
Department of Cellular and Molecular Medicine, Faculty of Health Sciences, University of Copenhagen, 3C Blegdamsvej, 2200, Copenhagen N, Denmark.
Biotech Research and Innovation Centre, University of Copenhagen, Ole Maaløes Vej 5, 2200, Copenhagen N, Denmark.
J Neuroinflammation. 2016 Mar 8;13(1):59. doi: 10.1186/s12974-016-0519-5.
Secretion of proteopathic α-synuclein (α-SNC) species from neurons is a suspected driving force in the propagation of Parkinson's disease (PD). We have previously implicated exophagy, the exocytosis of autophagosomes, as a dominant mechanism of α-SNC secretion in differentiated PC12 or SH-SY5Y nerve cells. Here we have examined the regulation of exophagy associated with different forms of nerve cell stress relevant to PD.
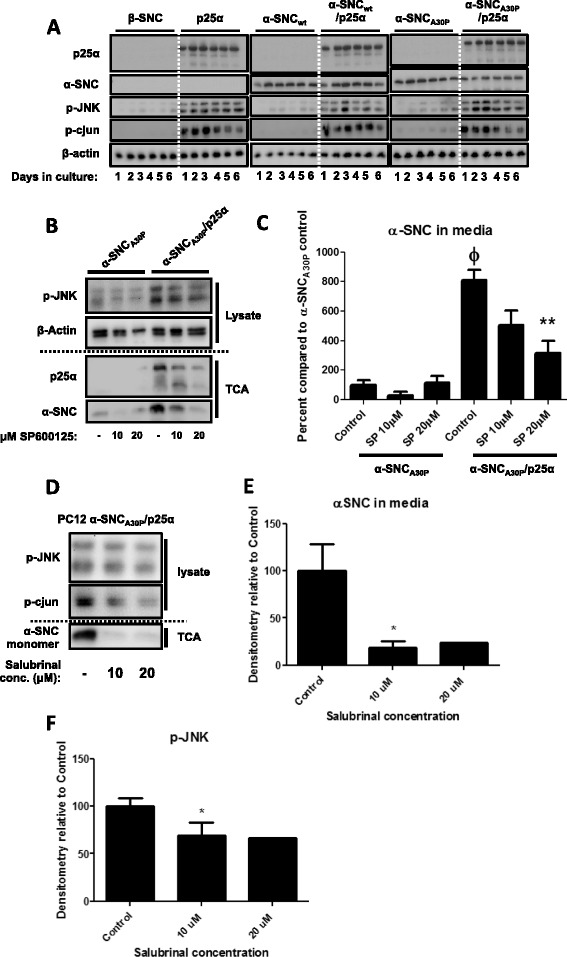
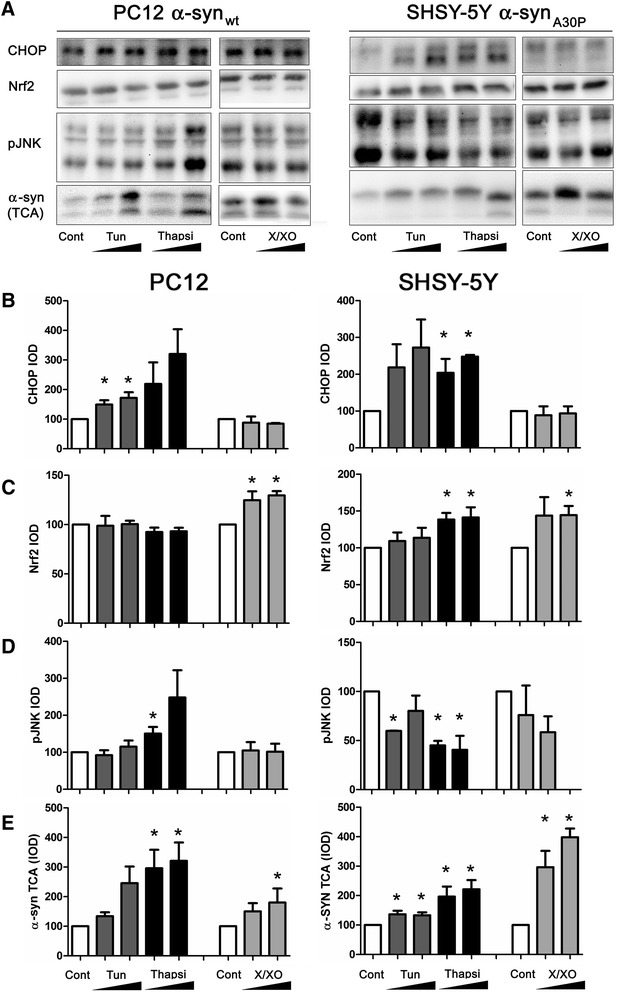
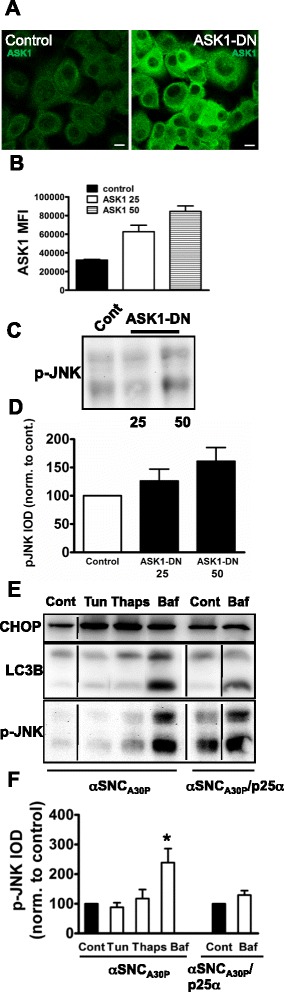
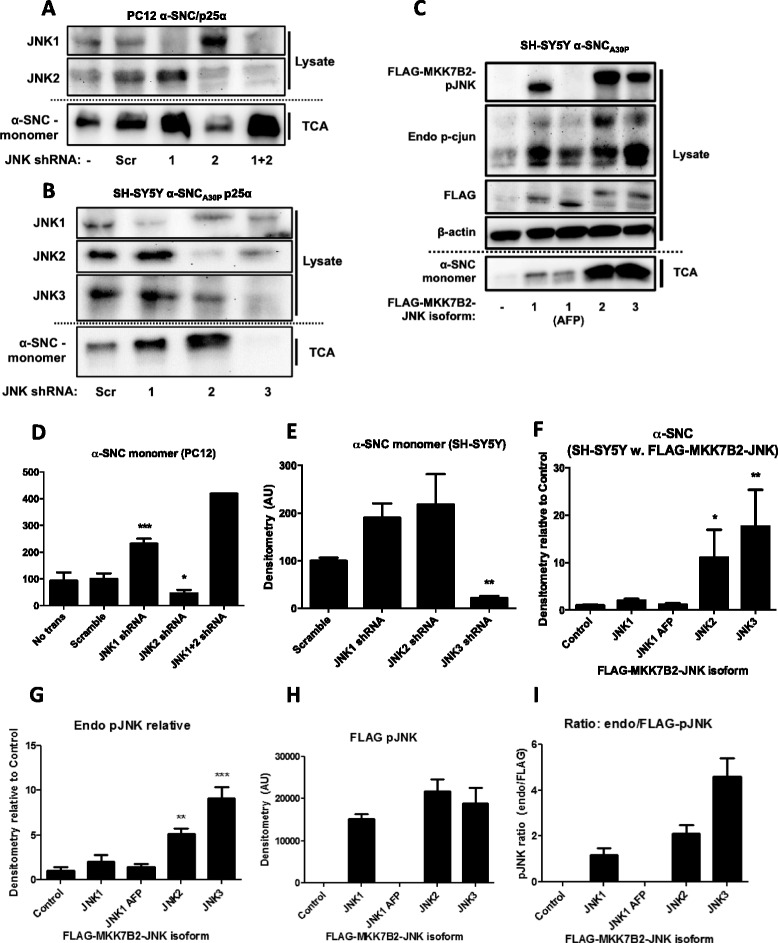
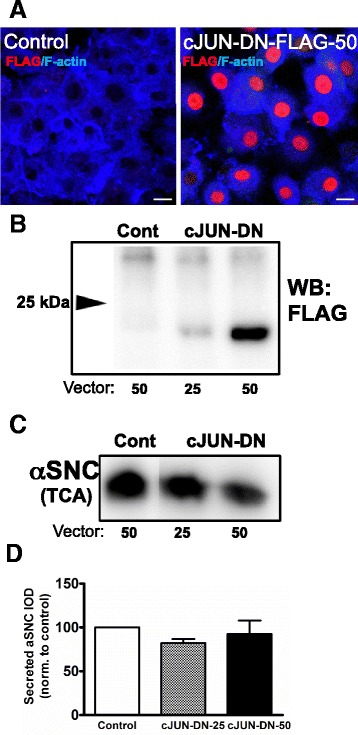
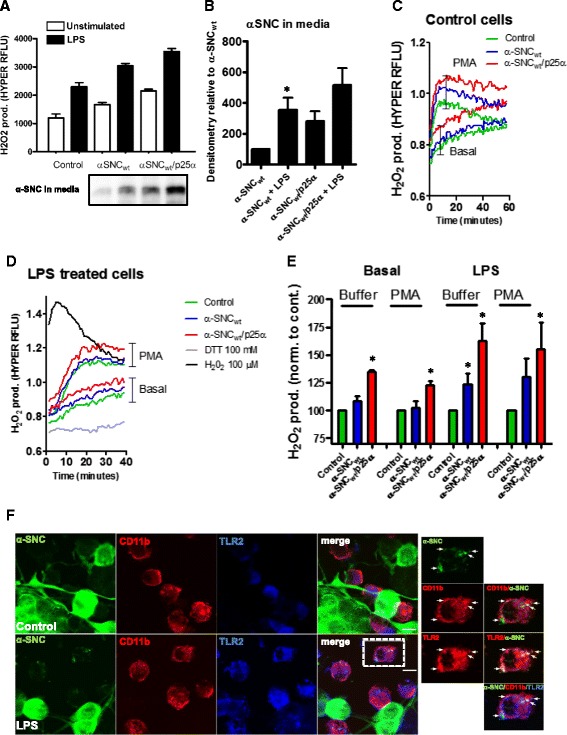
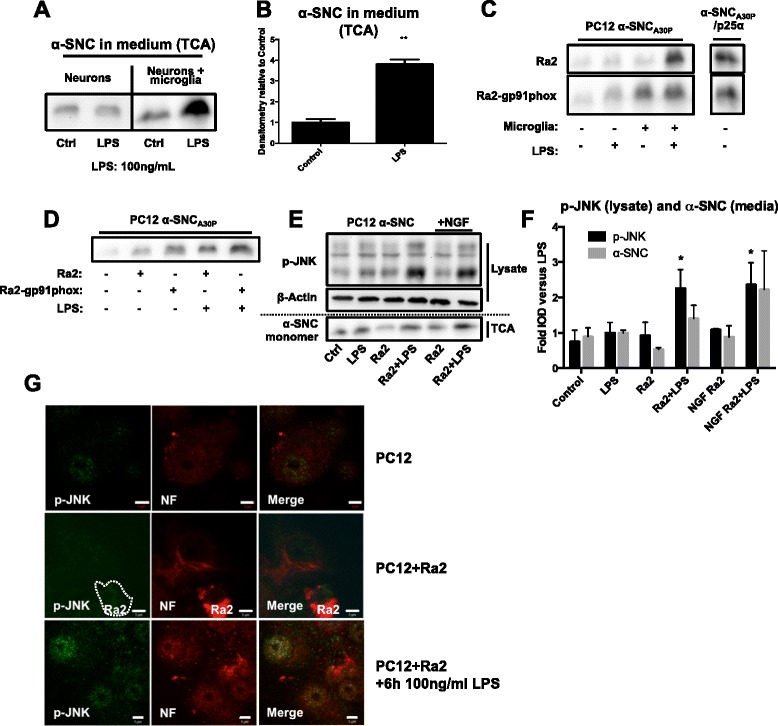
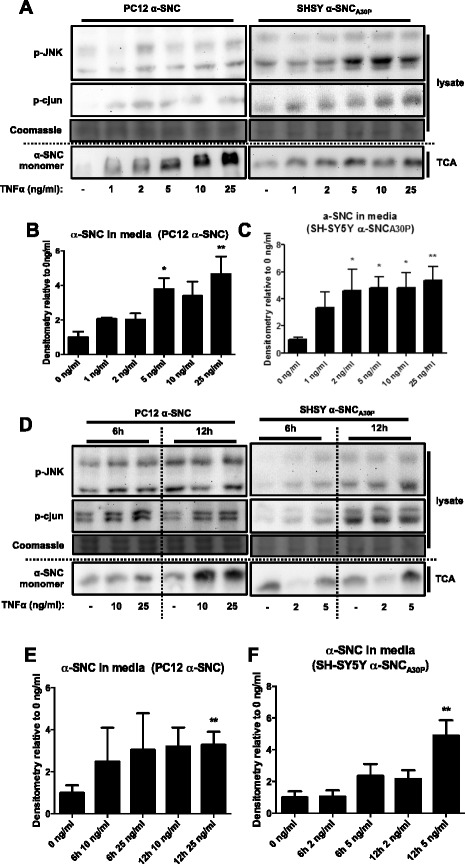
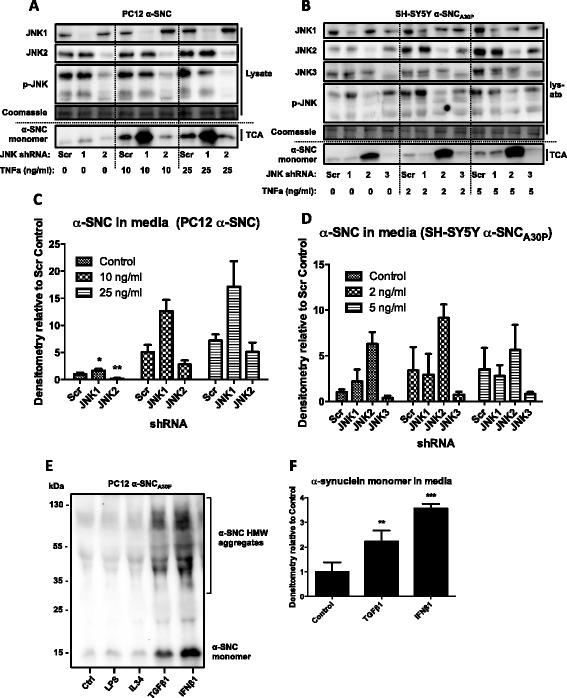
We identify cJUN-N-terminal kinase (JNK) activity as pivotal in the secretory fate of autophagosomes containing α-SNC. Pharmacological inhibition or genetic (shRNA) knockdown of JNK2 or JNK3 decreases α-SNC secretion in differentiated PC12 and SH-SY5Y cells, respectively. Conversely, expression of constitutively active mitogen-activated protein kinase kinase 7 (MKK7)-JNK2 and -JNK3 constructs augment secretion. The transcriptional activity of cJUN was not required for the observed effects. We establish a causal relationship between increased α-SNC release by exophagy and JNK activation subsequent to lysosomal fusion deficiency (overexpression of Lewy body-localized protein p25α or bafilomycin A1). JNK activation following neuronal ER or oxidative stress was not correlated with exophagy, but of note, we demonstrate that reciprocal signaling between microglia and neurons modulates α-SNC secretion. NADPH oxidase activity of microglia cell lines was upregulated by direct co-culture with α-SNC-expressing PC12 neurons or by passive transfer of nerve cell-conditioned medium. Conversely, inflammatory factors secreted from activated microglia increased JNK activation and α-SNC secretion several-fold in PC12 cells. While we do not identify these factors, we extend our observations by showing that exposure of neurons in monoculture to TNFα, a classical pro-inflammatory mediator of activated microglia, is sufficient to increase α-SNC secretion in a mechanism dependent on JNK2 or JNK3. In continuation hereof, we show that also IFNβ and TGFβ increase the release of α-SNC from PC12 neurons.
We implicate stress kinases of the JNK family in the regulation of exophagy and release of α-SNC following endogenous or exogenous stimulation. In a wider scope, our results imply that microglia not only inflict bystander damage to neurons in late phases of inflammatory brain disease but may also be active mediators of disease propagation.
神经元分泌致病性α-突触核蛋白(α-SNC)被怀疑是帕金森病(PD)传播的驱动力。我们之前认为外噬作用,即自噬体的胞吐作用,是分化的PC12或SH-SY5Y神经细胞中α-SNC分泌的主要机制。在此,我们研究了与PD相关的不同形式神经细胞应激相关的外噬作用的调节。
我们确定c-JUN氨基末端激酶(JNK)活性在含有α-SNC的自噬体的分泌命运中起关键作用。分别对JNK2或JNK3进行药理学抑制或基因(shRNA)敲低可降低分化的PC12和SH-SY5Y细胞中的α-SNC分泌。相反,组成型活性丝裂原活化蛋白激酶激酶7(MKK7)-JNK2和-JNK3构建体的表达增强了分泌。观察到的效应不需要c-JUN的转录活性。我们在内源性溶酶体融合缺陷(路易小体定位蛋白p25α过表达或巴佛洛霉素A1)后,建立了外噬作用增加α-SNC释放与JNK激活之间的因果关系。神经元内质网或氧化应激后的JNK激活与外噬作用无关,但值得注意的是,我们证明小胶质细胞与神经元之间的相互信号传导调节α-SNC分泌。小胶质细胞系的NADPH氧化酶活性通过与表达α-SNC的PC12神经元直接共培养或通过神经细胞条件培养基的被动转移而上调。相反,活化的小胶质细胞分泌的炎症因子使PC12细胞中的JNK激活和α-SNC分泌增加了几倍。虽然我们未确定这些因子,但我们通过显示将单培养中的神经元暴露于TNFα(一种活化小胶质细胞的经典促炎介质)足以通过依赖JNK2或JNK3的机制增加α-SNC分泌来扩展我们的观察结果。在此基础上,我们还表明IFNβ和TGFβ也增加了PC12神经元中α-SNC的释放。
我们认为JNK家族的应激激酶在内源性或外源性刺激后外噬作用和α-SNC释放的调节中起作用。从更广泛的范围来看,我们的结果意味着小胶质细胞不仅在炎症性脑疾病的晚期对神经元造成旁观者损伤,而且可能也是疾病传播的活跃介质。