Sarowar Tasnuva, Grabrucker Stefanie, Föhr Karl, Mangus Katharina, Eckert Matti, Bockmann Juergen, Boeckers Tobias M, Grabrucker Andreas M
WG Molecular Analysis of Synaptopathies, Neurology Department, Ulm University, Albert-Einstein-Allee 11, D-89081, Ulm, Germany.
Institute for Anatomy and Cell Biology, Ulm University, Albert-Einstein-Allee 11, D-89081, Ulm, Germany.
Mol Brain. 2016 Mar 11;9:28. doi: 10.1186/s13041-016-0206-6.
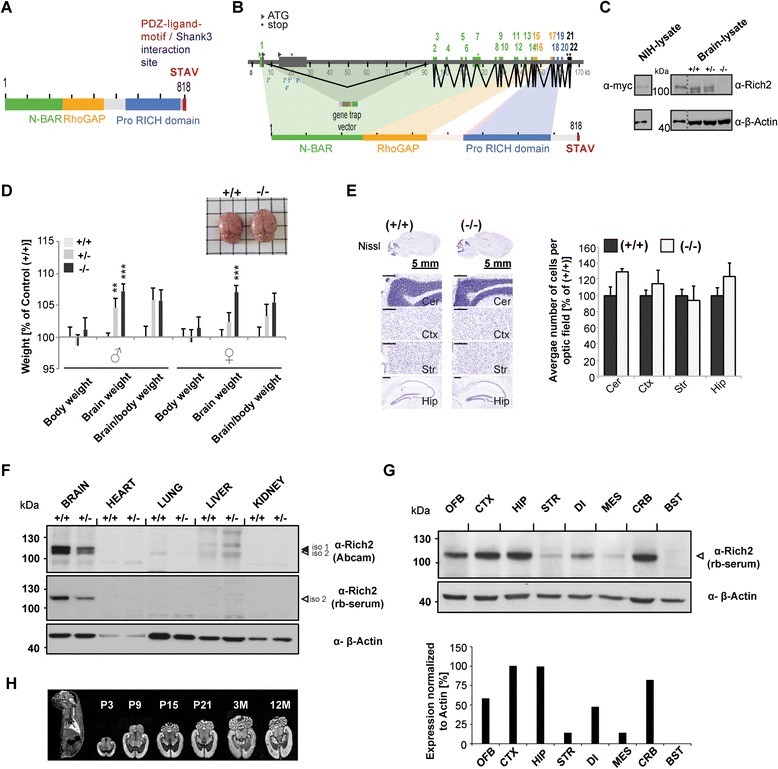
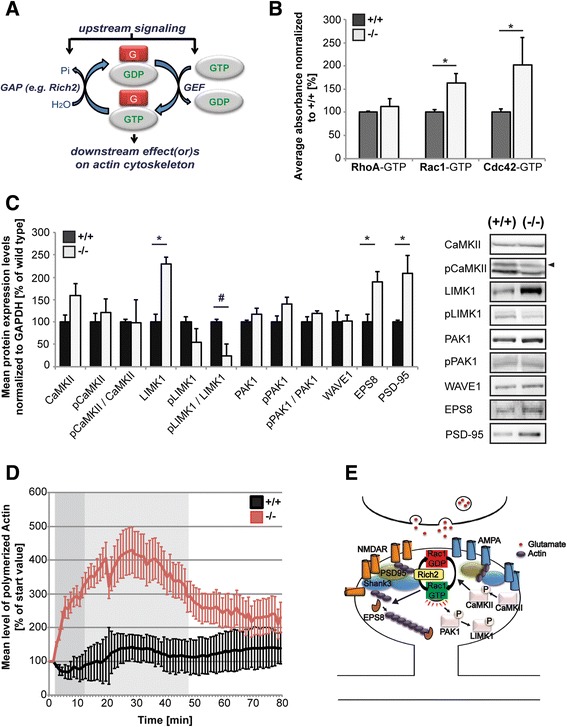
The majority of neurons within the central nervous system receive their excitatory inputs via small, actin-rich protrusions called dendritic spines. Spines can undergo rapid morphological alterations according to synaptic activity. This mechanism is implicated in learning and memory formation as it is ultimately altering the number and distribution of receptors and proteins at the post-synaptic membrane, thereby regulating synaptic input. The Rho-family GTPases play an important role in regulating this spine plasticity by the interaction with cytoskeletal components and several signaling pathways within the spine compartment. Rho-GAP interacting CIP4 homologue2/RICH2 is a Rho-GAP protein regulating small GTPases and was identified as an interaction partner of the scaffolding protein SHANK3 at post-synaptic densities.
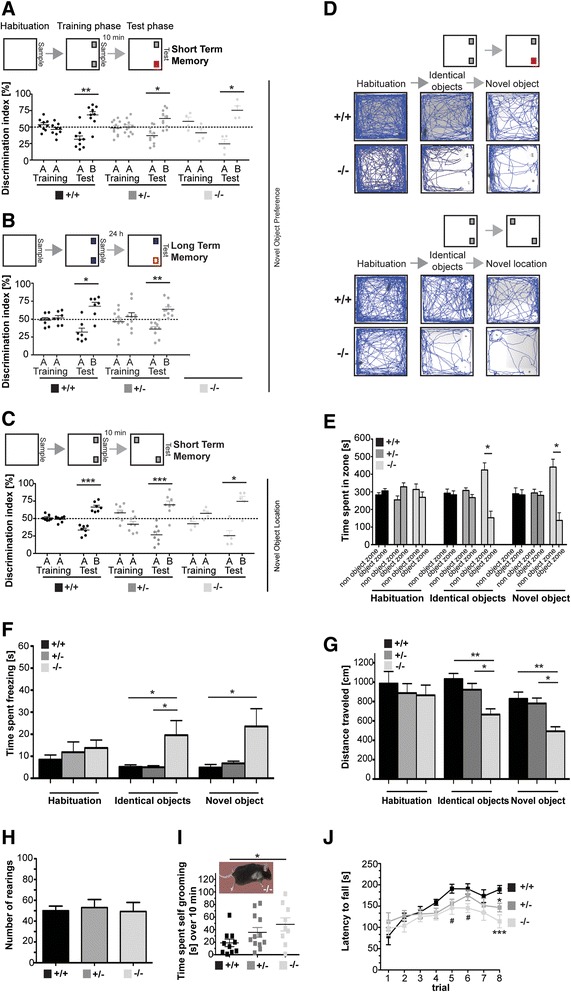
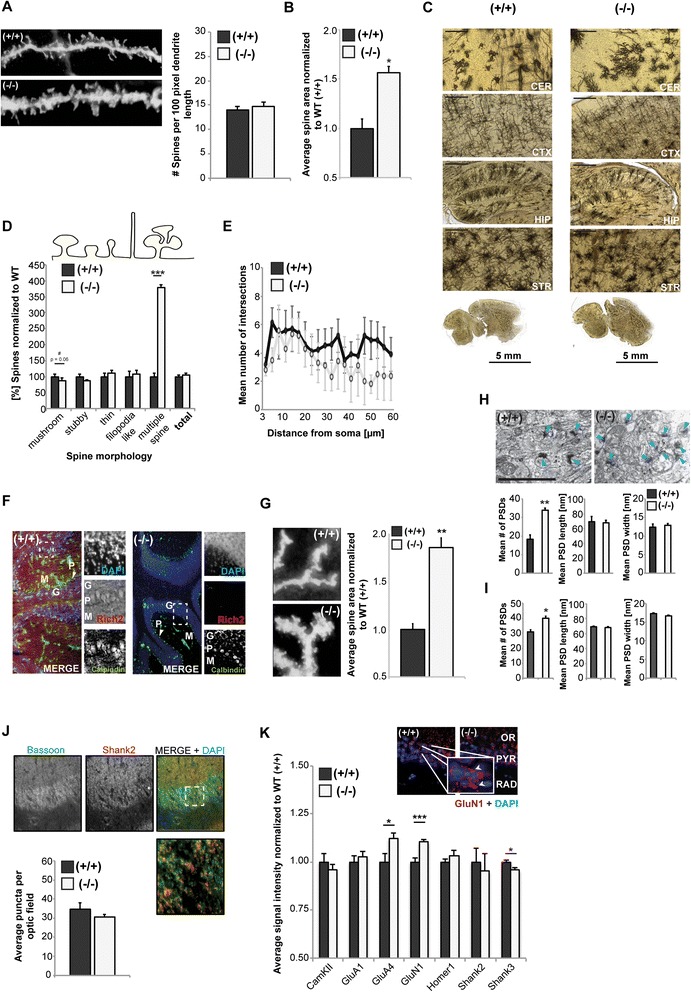
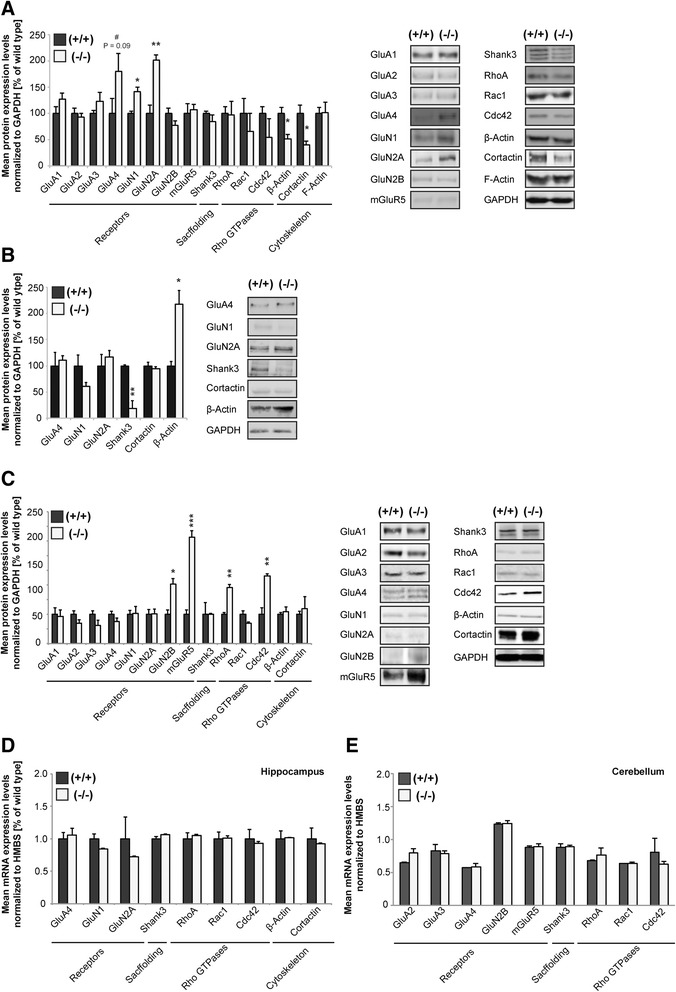
Here, we characterize the loss of RICH2 in a novel mouse model. Our results show that RICH2 KO animals display a selective and highly significant fear of novel objects and increased stereotypic behavior as well as impairment of motor learning. We found an increase in multiple spine synapses in the hippocampus and cerebellum along with alterations in receptor composition and actin polymerization. Furthermore, we observed that the loss of RICH2 leads to a disinhibition of synaptic RAC1 in vivo.
The results are in line with the reported role of RAC1 activity being essential for activity-dependent spine enlargement. Since SHANK3 mutations are known to be causative for neuropsychiatric diseases of the Autism Spectrum (ASD), a disintegrated SHANK3/RICH2 complex at synaptic sites might at least in part be responsible for abnormal spine formation and plasticity in ASDs.
中枢神经系统中的大多数神经元通过称为树突棘的富含肌动蛋白的小突起接收兴奋性输入。树突棘可根据突触活动迅速发生形态改变。这一机制与学习和记忆形成有关,因为它最终会改变突触后膜上受体和蛋白质的数量及分布,从而调节突触输入。Rho家族GTP酶通过与细胞骨架成分及树突棘区域内的多种信号通路相互作用,在调节这种树突棘可塑性方面发挥重要作用。Rho-GAP相互作用CIP4同源物2/RICH2是一种调节小GTP酶的Rho-GAP蛋白,被确定为突触后致密区支架蛋白SHANK3的相互作用伴侣。
在此,我们在一种新型小鼠模型中对RICH2缺失进行了表征。我们的结果表明,RICH2基因敲除动物对新物体表现出选择性且高度显著的恐惧,刻板行为增加,运动学习受损。我们发现海马体和小脑中多个棘突触增加,同时受体组成和肌动蛋白聚合发生改变。此外,我们观察到RICH2缺失导致体内突触RAC1去抑制。
这些结果与已报道的RAC1活性对依赖活动的树突棘增大至关重要的作用一致。由于已知SHANK3突变是自闭症谱系神经精神疾病的病因,突触部位SHANK3/RICH2复合物的解体可能至少部分导致了自闭症中异常的树突棘形成和可塑性。