Song Li, Sabunciyan Sarven, Florea Liliana
Center for Computational Biology, McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins School of Medicine, Baltimore, MD 21205, USA Department of Computer Science, Johns Hopkins University, Baltimore, MD 21218, USA.
Department of Pediatrics, Johns Hopkins School of Medicine, Baltimore, MD 21287, USA.
Nucleic Acids Res. 2016 Jun 2;44(10):e98. doi: 10.1093/nar/gkw158. Epub 2016 Mar 14.
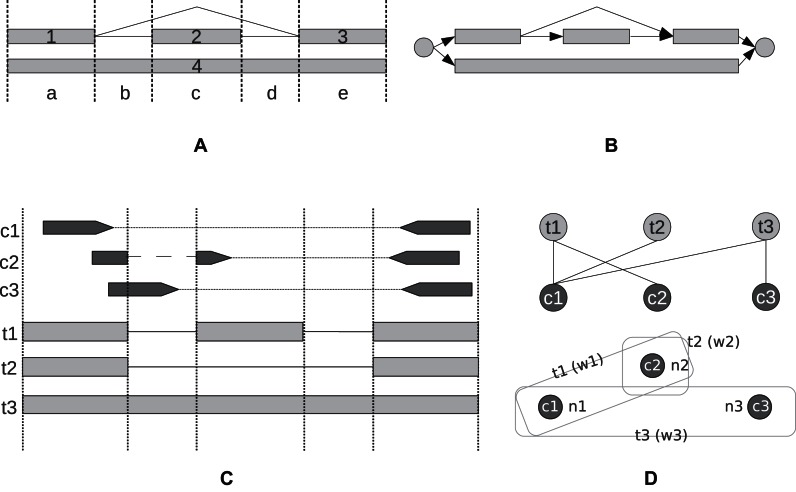
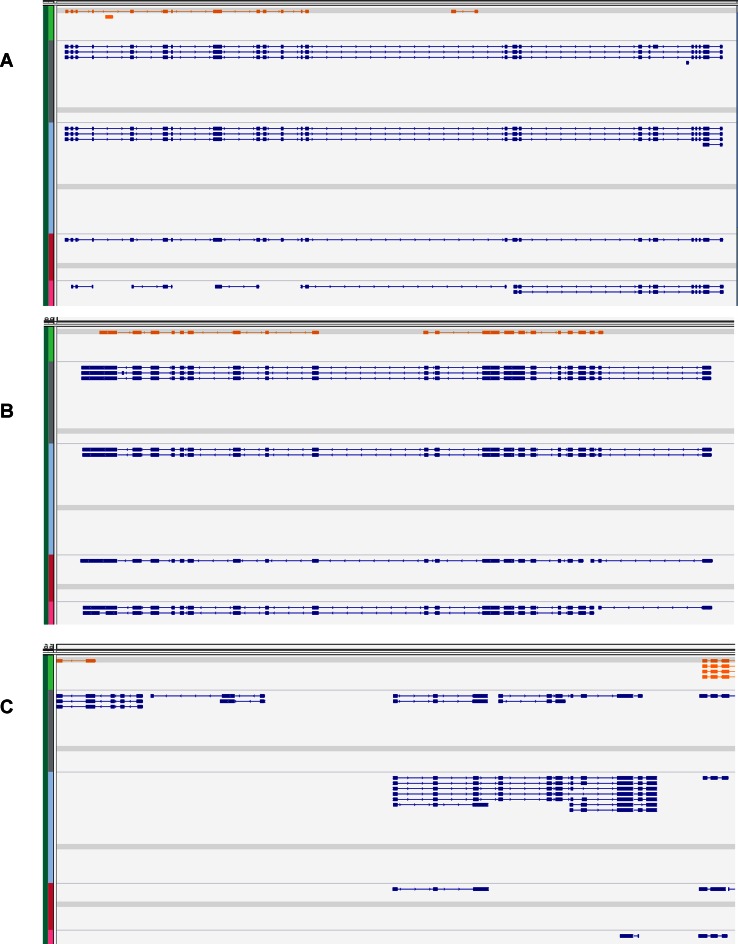
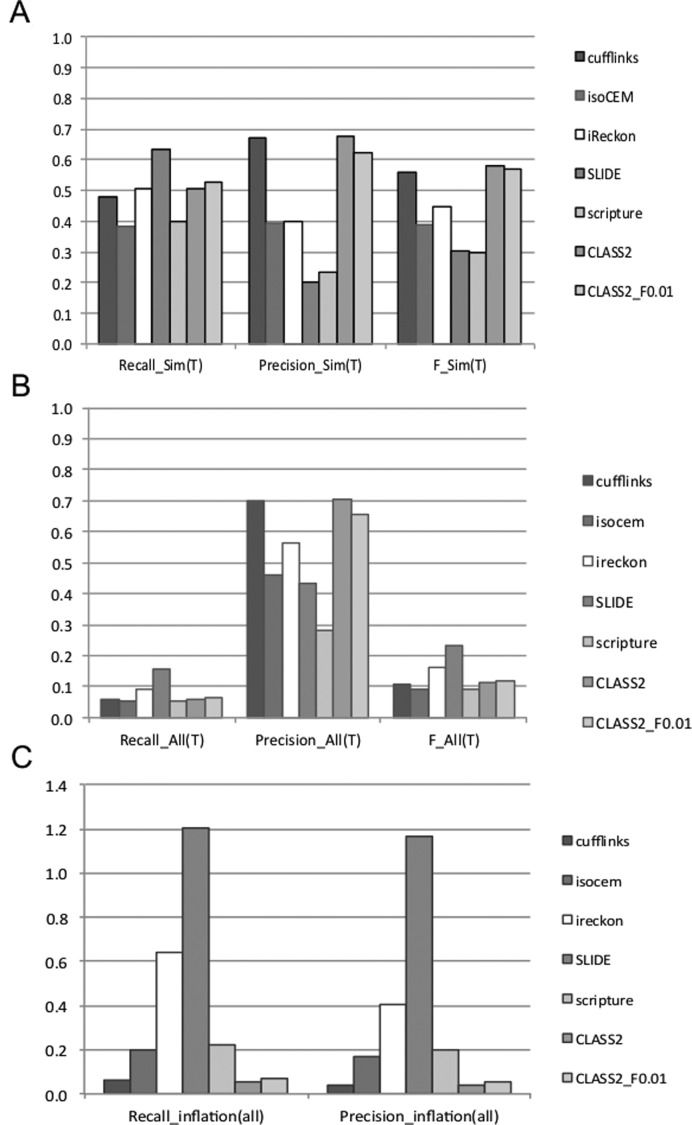
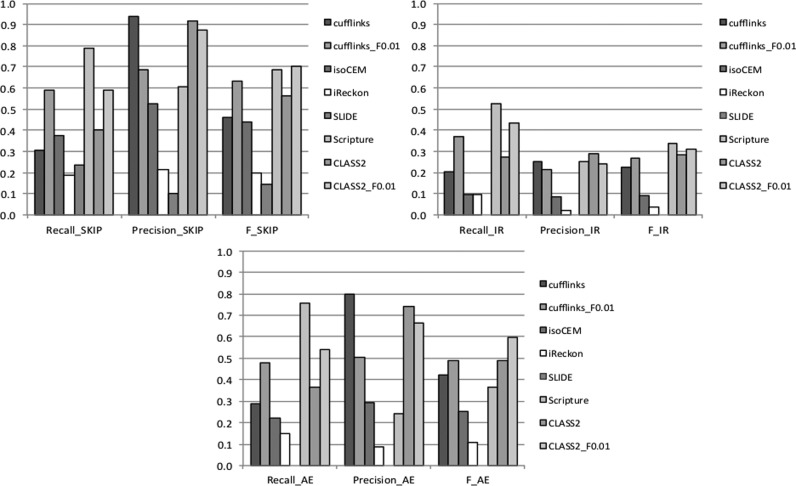
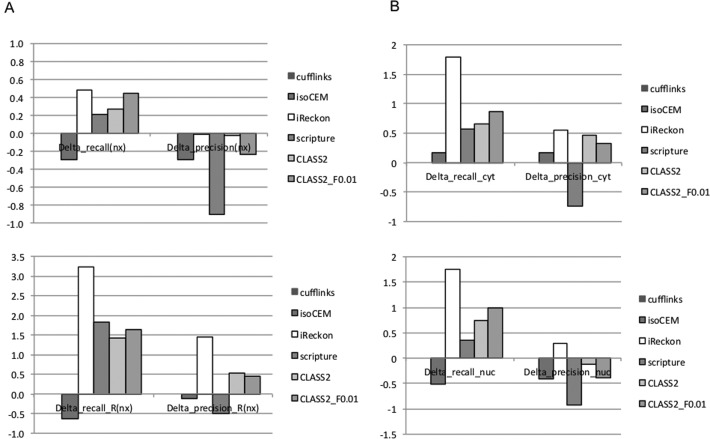
Next generation sequencing of cellular RNA is making it possible to characterize genes and alternative splicing in unprecedented detail. However, designing bioinformatics tools to accurately capture splicing variation has proven difficult. Current programs can find major isoforms of a gene but miss lower abundance variants, or are sensitive but imprecise. CLASS2 is a novel open source tool for accurate genome-guided transcriptome assembly from RNA-seq reads based on the model of splice graph. An extension of our program CLASS, CLASS2 jointly optimizes read patterns and the number of supporting reads to score and prioritize transcripts, implemented in a novel, scalable and efficient dynamic programming algorithm. When compared against reference programs, CLASS2 had the best overall accuracy and could detect up to twice as many splicing events with precision similar to the best reference program. Notably, it was the only tool to produce consistently reliable transcript models for a wide range of applications and sequencing strategies, including ribosomal RNA-depleted samples. Lightweight and multi-threaded, CLASS2 requires <3GB RAM and can analyze a 350 million read set within hours, and can be widely applied to transcriptomics studies ranging from clinical RNA sequencing, to alternative splicing analyses, and to the annotation of new genomes.
对细胞RNA进行下一代测序能够以前所未有的详细程度对基因和可变剪接进行表征。然而,事实证明设计能够准确捕捉剪接变异的生物信息学工具颇具难度。当前的程序能够找到基因的主要异构体,但会遗漏丰度较低的变体,或者虽灵敏但不够精确。CLASS2是一种新颖的开源工具,用于基于剪接图模型从RNA测序读数中进行准确的基因组引导转录组组装。作为我们的CLASS程序的扩展,CLASS2联合优化读数模式和支持读数的数量,以便对转录本进行评分和排序,这是通过一种新颖、可扩展且高效的动态规划算法实现的。与参考程序相比,CLASS2具有最佳的总体准确性,能够检测到的剪接事件数量最多可达最佳参考程序的两倍,且精度与之相似。值得注意的是,它是唯一一种能够为广泛的应用和测序策略(包括核糖体RNA去除样本)生成始终可靠的转录本模型的工具。CLASS2轻量级且支持多线程,所需内存小于3GB,能够在数小时内分析3.5亿个读数集,并且可广泛应用于从临床RNA测序到可变剪接分析以及新基因组注释等转录组学研究。