Szabo Emese, Schneider Hannah, Seystahl Katharina, Rushing Elisabeth Jane, Herting Frank, Weidner K Michael, Weller Michael
Laboratory of Molecular Neuro-Oncology, Department of Neurology (E.S., H.S., K.S., M.W.), and Institute of Neuropathology, University Hospital Zurich, Zurich, Switzerland (E.J.R); Roche Innovation Center Penzberg, Roche Pharma Research and Early Development, Nonnenwald 2, Penzberg D-82372, Germany (F.H., K.M.W.).
Laboratory of Molecular Neuro-Oncology, Department of Neurology (E.S., H.S., K.S., M.W.), and Institute of Neuropathology, University Hospital Zurich, Zurich, Switzerland (E.J.R); Roche Innovation Center Penzberg, Roche Pharma Research and Early Development, Nonnenwald 2, Penzberg D-82372, Germany (F.H., K.M.W.)
Neuro Oncol. 2016 Sep;18(9):1242-52. doi: 10.1093/neuonc/now043. Epub 2016 Mar 23.
Although the vascular endothelial growth factor (VEGF)/VEGF receptor (VEGFR) system has become a prime target for antiangiogenic treatment, its biological role in glioblastoma beyond angiogenesis has remained controversial.
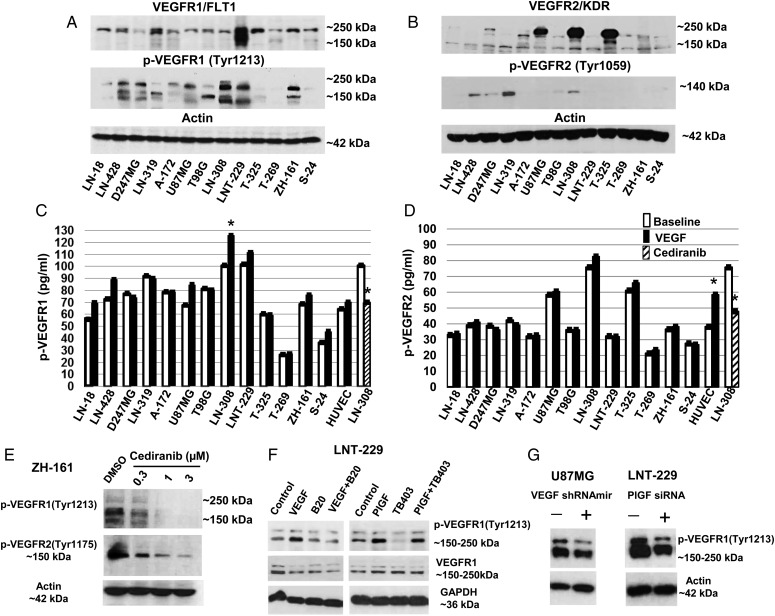
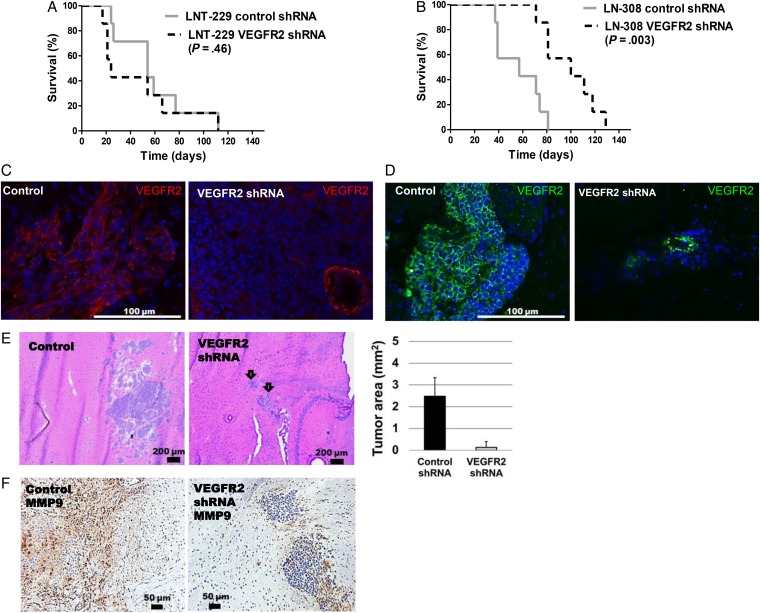
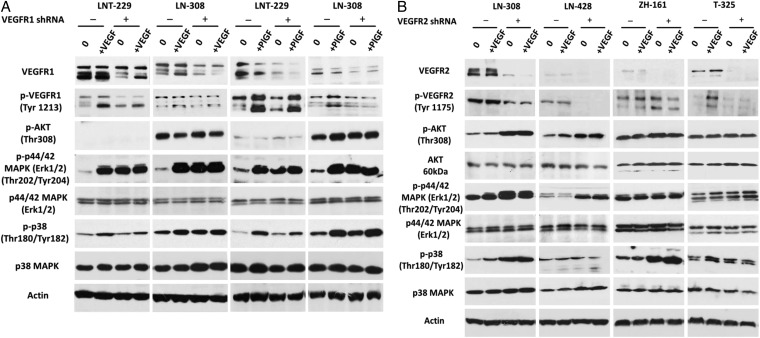
Using neutralizing antibodies to VEGF or placental growth factor (PlGF) or the tyrosine kinase inhibitor, cediranib, or lentiviral gene silencing, we delineated autocrine signaling in glioma cell lines. The in vivo effects of VEGFR1 and VEGFR2 depletion were evaluated in orthotopic glioma xenograft models.
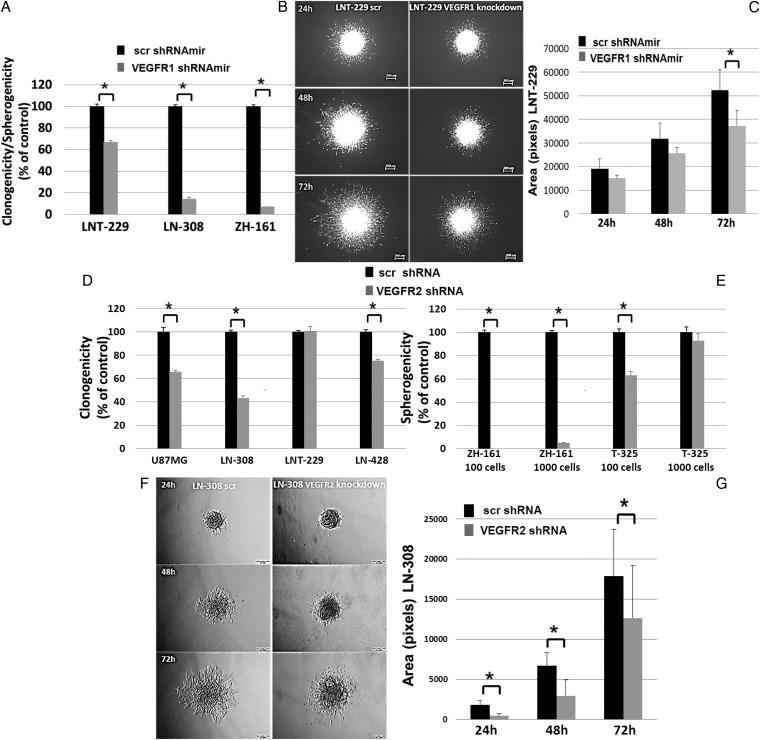
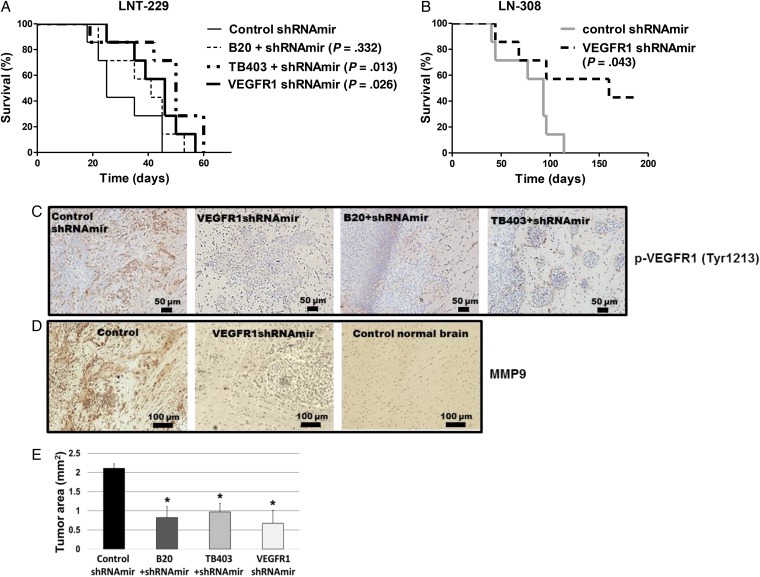
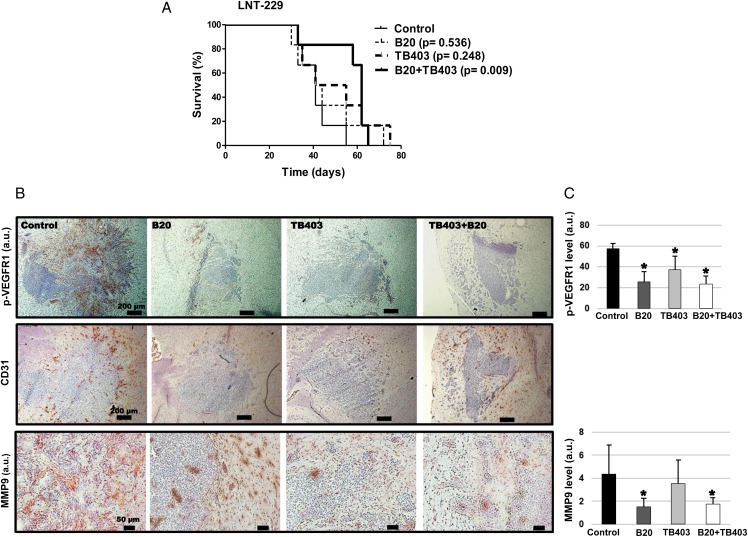
VEGFR1 and VEGFR2 modulated glioma cell clonogenicity, viability, and invasiveness in vitro in an autocrine, cell-line-specific manner. VEGFR1 silencing promoted mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) signaling, whereas VEGFR2 silencing resulted in cell-type dependent activation of the protein kinase B (PKB)/AKT and MAPK/ERK pathways. These responses may represent specific escape mechanisms from VEGFR inhibition. The survival of orthotopic glioma-bearing mice was prolonged upon VEGFR1 silencing in the LNT-229, LN-308, and U87MG models and upon VEGFR2 silencing in LN-308 and U87MG. Disruption of VEGFR1 and VEGFR2 signaling was associated with decreased tumor size, increased tumor necrosis, or loss of matrix metalloproteinase 9 (MMP9) immunoreactivity. Neutralizing VEGF and PlGF by specific antibodies was superior to either antibody treatment alone in the VEGFR1-dependent LNT-229 model.
Differential dependence on autocrine signaling through VEGFR1 and VEGFR2 suggests a need for biomarker-stratified VEGF(R)-based therapeutic approaches to glioblastoma.
尽管血管内皮生长因子(VEGF)/VEGF受体(VEGFR)系统已成为抗血管生成治疗的主要靶点,但其在胶质母细胞瘤中除血管生成外的生物学作用仍存在争议。
我们使用针对VEGF或胎盘生长因子(PlGF)的中和抗体、酪氨酸激酶抑制剂西地尼布或慢病毒基因沉默,来描绘胶质瘤细胞系中的自分泌信号传导。在原位胶质瘤异种移植模型中评估VEGFR1和VEGFR2缺失的体内效应。
VEGFR1和VEGFR2以自分泌、细胞系特异性方式在体外调节胶质瘤细胞的克隆形成能力、活力和侵袭性。VEGFR1沉默促进丝裂原活化蛋白激酶/细胞外信号调节激酶(MAPK/ERK)信号传导,而VEGFR2沉默导致蛋白激酶B(PKB)/AKT和MAPK/ERK途径的细胞类型依赖性激活。这些反应可能代表了VEGFR抑制的特定逃逸机制。在LNT - 229、LN - 308和U87MG模型中,VEGFR1沉默可延长原位荷胶质瘤小鼠的生存期,在LN - 308和U87MG模型中,VEGFR2沉默也可延长生存期。VEGFR1和VEGFR2信号传导的破坏与肿瘤大小减小、肿瘤坏死增加或基质金属蛋白酶9(MMP9)免疫反应性丧失有关。在VEGFR1依赖性LNT - 229模型中,用特异性抗体中和VEGF和PlGF优于单独使用任何一种抗体治疗。
对通过VEGFR1和VEGFR2的自分泌信号传导的差异依赖性表明,需要基于生物标志物分层的VEGF(R)治疗胶质母细胞瘤的方法。