Zhong Kaiyin, Karssen Lennart C, Kayser Manfred, Liu Fan
Department of Genetic Identification, Erasmus University Medical Center Rotterdam, Rotterdam, The Netherlands.
PolyOmica, Groningen, The Netherlands.
BMC Bioinformatics. 2016 Apr 8;17:156. doi: 10.1186/s12859-016-1006-9.
Compound Heterozygosity (CH) in classical genetics is the presence of two different recessive mutations at a particular gene locus. A relaxed form of CH alleles may account for an essential proportion of the missing heritability, i.e. heritability of phenotypes so far not accounted for by single genetic variants. Methods to detect CH-like effects in genome-wide association studies (GWAS) may facilitate explaining the missing heritability, but to our knowledge no viable software tools for this purpose are currently available.
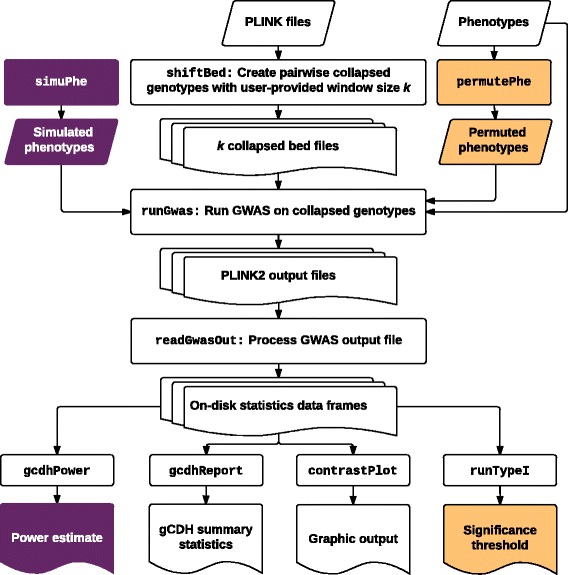
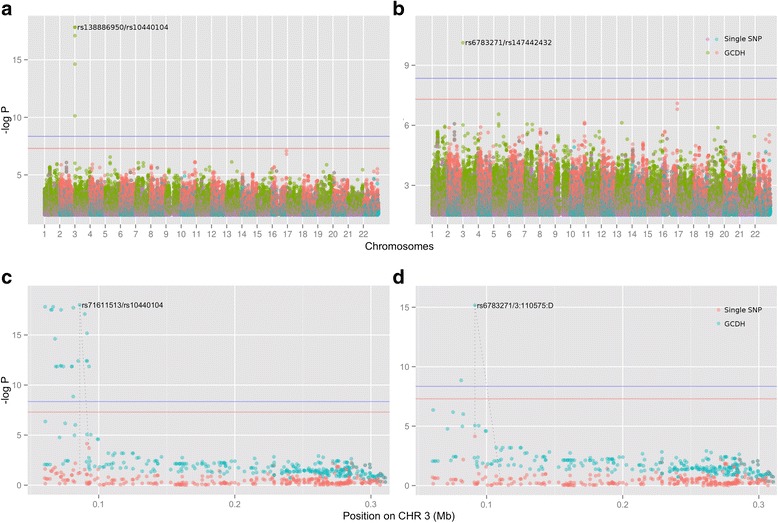
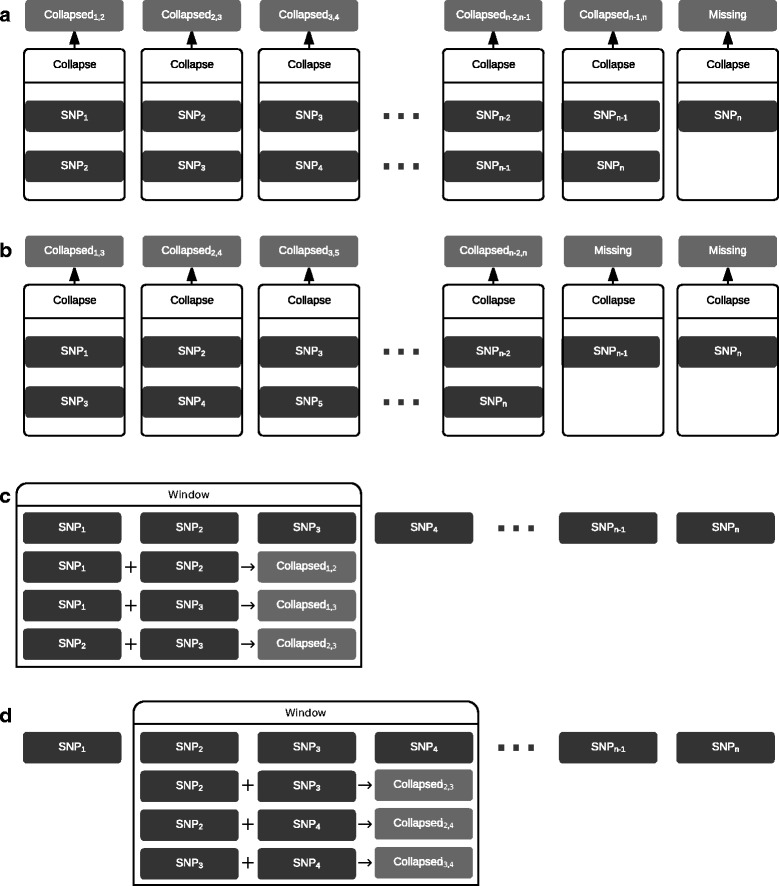
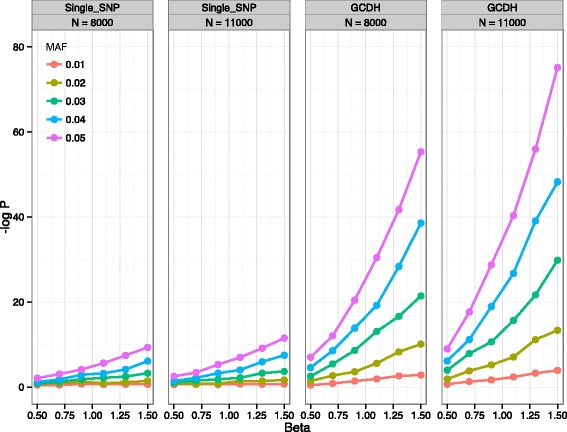
In this work we present the Generalized Compound Double Heterozygosity (GCDH) test and its implementation in the R package CollapsABEL. Time-consuming procedures are optimized for computational efficiency using Java or C++. Intermediate results are stored either in an SQL database or in a so-called big.matrix file to achieve reasonable memory footprint. Our large scale simulation studies show that GCDH is capable of discovering genetic associations due to CH-like interactions with much higher power than a conventional single-SNP approach under various settings, whether the causal genetic variations are available or not. CollapsABEL provides a user-friendly pipeline for genotype collapsing, statistical testing, power estimation, type I error control and graphics generation in the R language.
CollapsABEL provides a computationally efficient solution for screening general forms of CH alleles in densely imputed microarray or whole genome sequencing datasets. The GCDH test provides an improved power over single-SNP based methods in detecting the prevalence of CH in human complex phenotypes, offering an opportunity for tackling the missing heritability problem. Binary and source packages of CollapsABEL are available on CRAN ( https://cran.r-project.org/web/packages/CollapsABEL ) and the website of the GenABEL project ( http://www.genabel.org/packages ).
在经典遗传学中,复合杂合性(CH)是指在特定基因座上存在两个不同的隐性突变。CH等位基因的一种宽松形式可能占缺失遗传力的很大一部分,即迄今为止尚未由单个基因变异解释的表型遗传力。在全基因组关联研究(GWAS)中检测类似CH效应的方法可能有助于解释缺失的遗传力,但据我们所知,目前尚无适用于此目的的可行软件工具。
在这项工作中,我们提出了广义复合双杂合性(GCDH)检验及其在R包CollapsABEL中的实现。使用Java或C++对耗时的程序进行了优化以提高计算效率。中间结果存储在SQL数据库或所谓的big.matrix文件中,以实现合理的内存占用。我们的大规模模拟研究表明,无论因果基因变异是否可用,在各种设置下,GCDH都能够以比传统单核苷酸多态性(SNP)方法更高的功效发现由于类似CH相互作用而产生的基因关联。CollapsABEL提供了一个用户友好的管道,用于在R语言中进行基因型压缩、统计检验、功效估计、I型错误控制和图形生成。
CollapsABEL为在密集填充的微阵列或全基因组测序数据集中筛选CH等位基因的一般形式提供了一种计算高效的解决方案。GCDH检验在检测人类复杂表型中CH的普遍性方面比基于单SNP的方法具有更高的功效,为解决缺失遗传力问题提供了一个机会。CollapsABEL的二进制和源包可在CRAN(https://cran.r-project.org/web/packages/CollapsABEL)和GenABEL项目网站(http://www.genabel.org/packages)上获得。