Department of Agricultural and Environmental Biology, Graduate School of Agricultural and Life Sciences, The University of Tokyo, Tokyo, Japan.
PLoS Comput Biol. 2020 Feb 14;16(2):e1007663. doi: 10.1371/journal.pcbi.1007663. eCollection 2020 Feb.
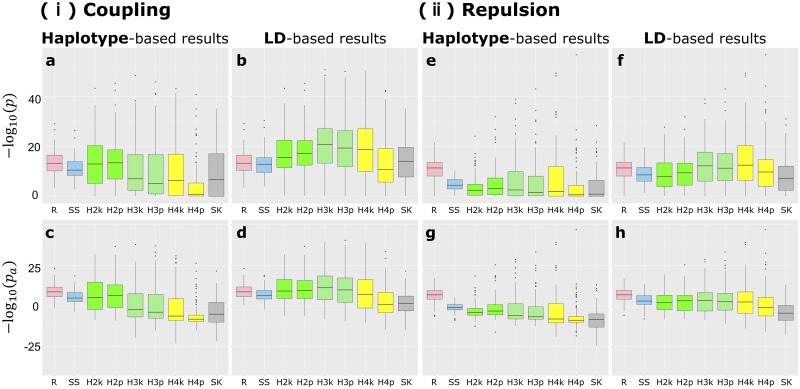
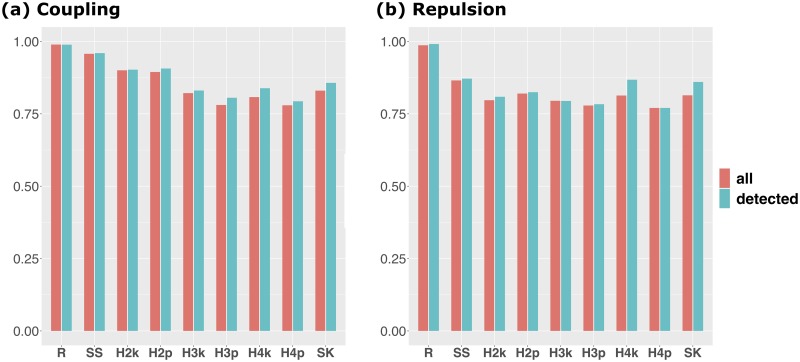
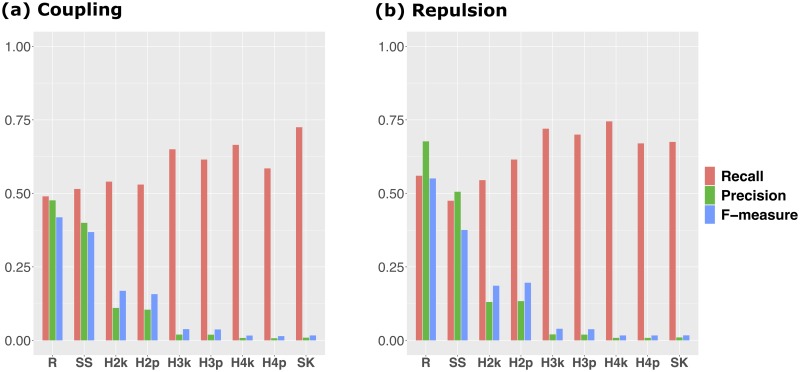
Difficulty in detecting rare variants is one of the problems in conventional genome-wide association studies (GWAS). The problem is closely related to the complex gene compositions comprising multiple alleles, such as haplotypes. Several single nucleotide polymorphism (SNP) set approaches have been proposed to solve this problem. These methods, however, have been rarely discussed in connection with haplotypes. In this study, we developed a novel SNP-set method named "RAINBOW" and applied the method to haplotype-based GWAS by regarding a haplotype block as a SNP-set. Combining haplotype block estimation and SNP-set GWAS, haplotype-based GWAS can be conducted without prior information of haplotypes. We prepared 100 datasets of simulated phenotypic data and real marker genotype data of Oryza sativa subsp. indica, and performed GWAS of the datasets. We compared the power of our method, the conventional single-SNP GWAS, the conventional haplotype-based GWAS, and the conventional SNP-set GWAS. Our proposed method was shown to be superior to these in three aspects: (1) controlling false positives; (2) in detecting causal variants without relying on the linkage disequilibrium if causal variants were genotyped in the dataset; and (3) it showed greater power than the other methods, i.e., it was able to detect causal variants that were not detected by the others, primarily when the causal variants were located very close to each other, and the directions of their effects were opposite. By using the SNP-set approach as in this study, we expect that detecting not only rare variants but also genes with complex mechanisms, such as genes with multiple causal variants, can be realized. RAINBOW was implemented as an R package named "RAINBOWR" and is available from CRAN (https://cran.r-project.org/web/packages/RAINBOWR/index.html) and GitHub (https://github.com/KosukeHamazaki/RAINBOWR).
在传统的全基因组关联研究(GWAS)中,检测罕见变异是一个问题。这个问题与包含多个等位基因(如单倍型)的复杂基因组成密切相关。已经提出了几种单核苷酸多态性(SNP)集方法来解决这个问题。然而,这些方法很少与单倍型相关联讨论。在这项研究中,我们开发了一种名为“RAINBOW”的新 SNP 集方法,并通过将单倍型块视为 SNP 集将该方法应用于基于单倍型的 GWAS。通过组合单倍型块估计和 SNP 集 GWAS,可以在没有单倍型先验信息的情况下进行基于单倍型的 GWAS。我们准备了 100 个模拟表型数据和 Oryza sativa subsp. indica 的真实标记基因型数据集,并对这些数据集进行了 GWAS。我们比较了我们的方法、传统的单 SNP GWAS、传统的基于单倍型的 GWAS 和传统的 SNP 集 GWAS 的功效。我们的方法在三个方面表现出优越性:(1)控制假阳性;(2)在不依赖连锁不平衡的情况下检测因果变异,如果因果变异在数据集中被基因分型;(3)它比其他方法具有更大的功效,即能够检测到其他方法无法检测到的因果变异,主要是当因果变异彼此非常接近且它们的作用方向相反时。通过在这项研究中使用 SNP 集方法,我们期望不仅可以检测罕见变异,还可以检测具有复杂机制的基因,例如具有多个因果变异的基因。RAINBOW 被实现为一个名为“RAINBOWR”的 R 包,并可从 CRAN(https://cran.r-project.org/web/packages/RAINBOWR/index.html)和 GitHub(https://github.com/KosukeHamazaki/RAINBOWR)获得。