Akinola Richard O, Mazandu Gaston K, Mulder Nicola J
Computational Biology Group, Department of Integrative Biomedical Sciences, Medical School, Institute of Infectious Disease and Molecular Medicine, University of Cape TownCape Town, South Africa; Department of Mathematics, Faculty of Natural Sciences, University of JosJos, Nigeria.
Computational Biology Group, Department of Integrative Biomedical Sciences, Medical School, Institute of Infectious Disease and Molecular Medicine, University of Cape TownCape Town, South Africa; African Institute for Mathematical SciencesCape Town, South Africa; African Institute for Mathematical SciencesCape Coast, Ghana.
Front Genet. 2016 Mar 29;7:39. doi: 10.3389/fgene.2016.00039. eCollection 2016.
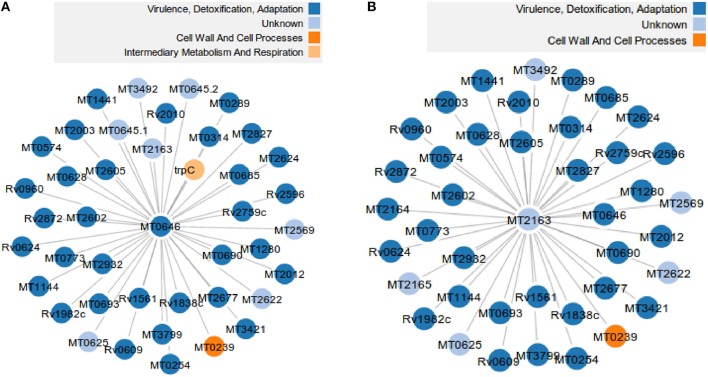
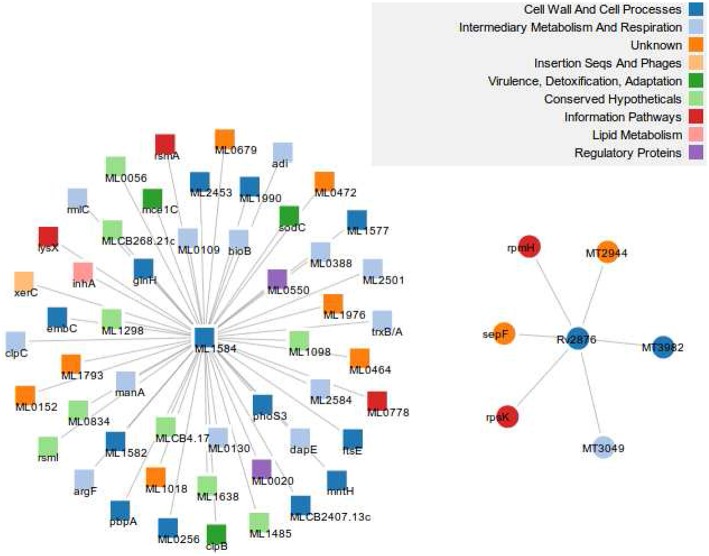
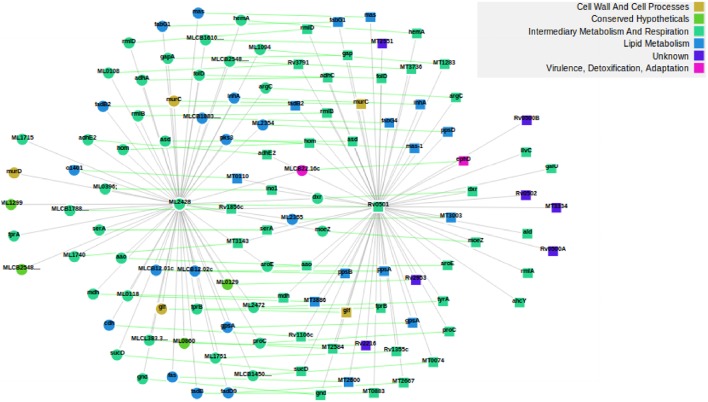
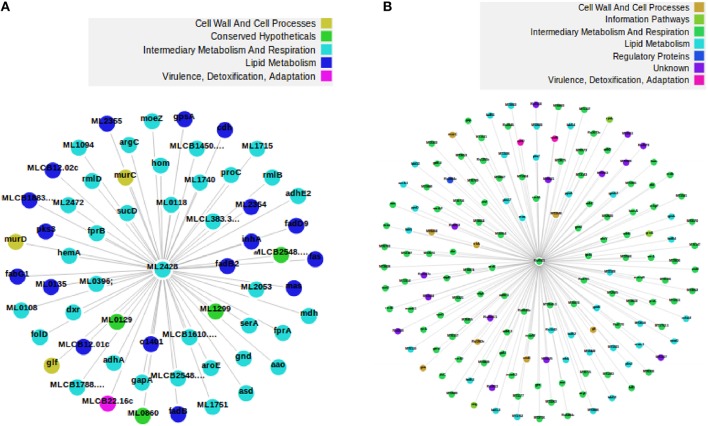
The advance in high-throughput sequencing technologies has yielded complete genome sequences of several organisms, including complete bacterial genomes. The growing number of these available sequenced genomes has enabled analyses of their dynamics, as well as the molecular and evolutionary processes which these organisms are under. Comparative genomics of different bacterial genomes have highlighted their genome size and gene content in association with lifestyles and adaptation to various environments and have contributed to enhancing our understanding of the mechanisms of their evolution. Protein-protein functional interactions mediate many essential processes for maintaining the stability of the biological systems under changing environmental conditions. Thus, these interactions play crucial roles in the evolutionary processes of different organisms, especially for obligate intracellular bacteria, proven to generally have reduced genome sizes compared to their nearest free-living relatives. In this study, we used the approach based on the Renormalization Group (RG) analysis technique and the Maximum-Excluded-Mass-Burning (MEMB) model to investigate the evolutionary process of genome reduction in relation to the organization of functional networks of two organisms. Using a Mycobacterium leprae (MLP) network in comparison with a Mycobacterium tuberculosis (MTB) network as a case study, we show that reductive evolution in MLP was as a result of removal of important proteins from neighbors of corresponding orthologous MTB proteins. While each orthologous MTB protein had an increase in number of interacting partners in most instances, the corresponding MLP protein had lost some of them. This work provides a quantitative model for mapping reductive evolution and protein-protein functional interaction network organization in terms of roles played by different proteins in the network structure.
高通量测序技术的进步已产生了几种生物的完整基因组序列,包括完整的细菌基因组。这些可用的已测序基因组数量不断增加,使得对其动态变化以及这些生物所经历的分子和进化过程进行分析成为可能。不同细菌基因组的比较基因组学突出了它们的基因组大小和基因含量与生活方式以及对各种环境的适应性之间的关系,并有助于增进我们对其进化机制的理解。蛋白质 - 蛋白质功能相互作用介导了在不断变化的环境条件下维持生物系统稳定性的许多基本过程。因此,这些相互作用在不同生物的进化过程中起着关键作用,特别是对于专性胞内细菌而言,事实证明它们的基因组大小通常比其最接近的自由生活亲属要小。在本研究中,我们使用基于重整化群(RG)分析技术和最大排除质量燃烧(MEMB)模型的方法,来研究与两种生物的功能网络组织相关的基因组缩减的进化过程。以麻风分枝杆菌(MLP)网络与结核分枝杆菌(MTB)网络进行比较作为案例研究,我们表明MLP中的简化进化是由于从相应直系同源MTB蛋白的邻居中去除了重要蛋白质所致。虽然在大多数情况下,每个直系同源MTB蛋白的相互作用伙伴数量有所增加,但相应的MLP蛋白却失去了其中一些伙伴。这项工作提供了一个定量模型,用于根据不同蛋白质在网络结构中所起的作用来描绘简化进化和蛋白质 - 蛋白质功能相互作用网络组织。