Alston Charlotte L, Howard Caoimhe, Oláhová Monika, Hardy Steven A, He Langping, Murray Philip G, O'Sullivan Siobhan, Doherty Gary, Shield Julian P H, Hargreaves Iain P, Monavari Ardeshir A, Knerr Ina, McCarthy Peter, Morris Andrew A M, Thorburn David R, Prokisch Holger, Clayton Peter E, McFarland Robert, Hughes Joanne, Crushell Ellen, Taylor Robert W
Wellcome Trust Centre for Mitochondrial Research, Institute of Neuroscience, Newcastle University, Newcastle upon Tyne, UK.
National Centre for Inherited Metabolic Disorders, Temple Street Children's University Hospital, Dublin, Ireland.
J Med Genet. 2016 Sep;53(9):634-41. doi: 10.1136/jmedgenet-2015-103576. Epub 2016 Apr 18.
Isolated Complex I deficiency is the most common paediatric mitochondrial disease presentation, associated with poor prognosis and high mortality. Complex I comprises 44 structural subunits with at least 10 ancillary proteins; mutations in 29 of these have so far been associated with mitochondrial disease but there are limited genotype-phenotype correlations to guide clinicians to the correct genetic diagnosis.
Patients were analysed by whole-exome sequencing, targeted capture or candidate gene sequencing. Clinical phenotyping of affected individuals was performed.
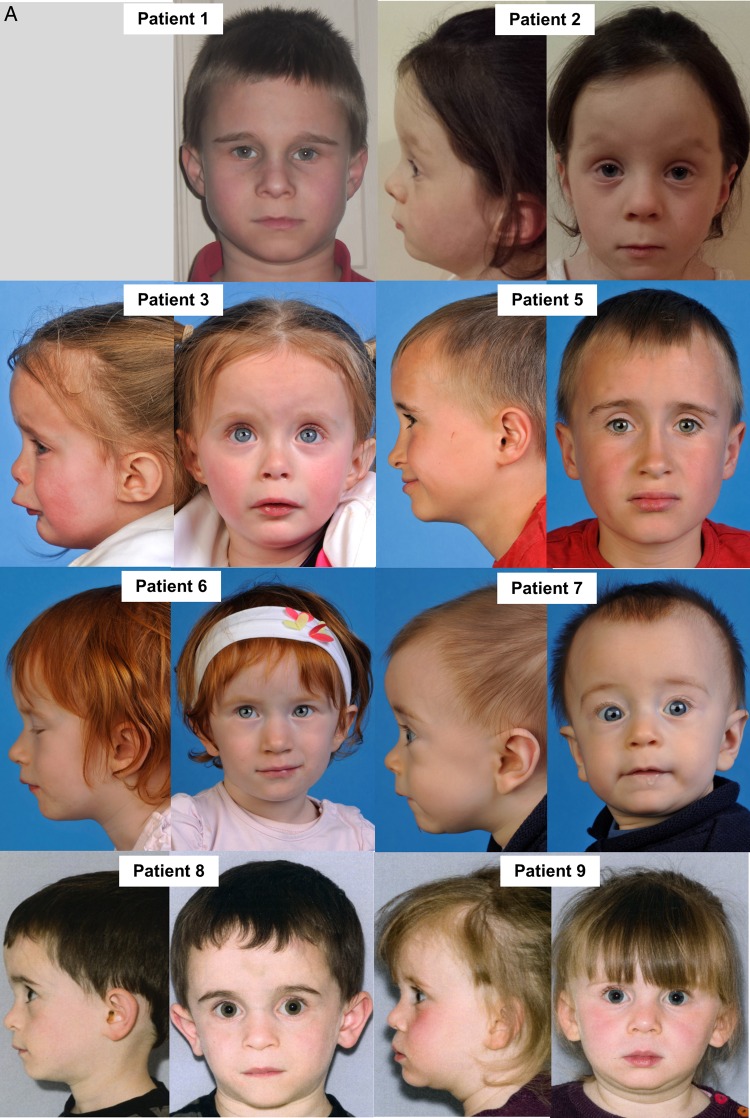
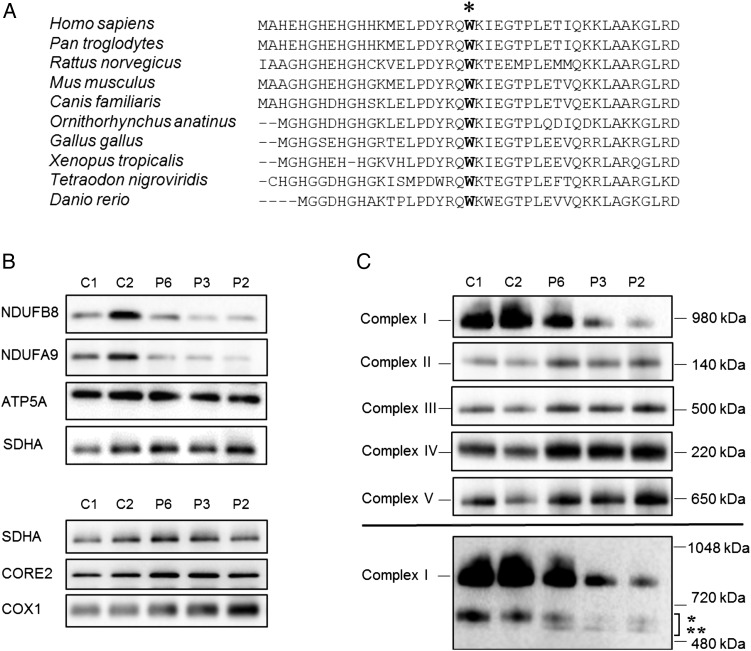
We identified a cohort of 10 patients from 8 families (7 families are of unrelated Irish ancestry) all of whom have short stature (<9th centile) and similar facial features including a prominent forehead, smooth philtrum and deep-set eyes associated with a recurrent homozygous c.64T>C, p.Trp22Arg NDUFB3 variant. Two sibs presented with primary short stature without obvious metabolic dysfunction. Analysis of skeletal muscle from three patients confirmed a defect in Complex I assembly.
Our report highlights that the long-term prognosis related to the p.Trp22Arg NDUFB3 mutation can be good, even for some patients presenting in acute metabolic crisis with evidence of an isolated Complex I deficiency in muscle. Recognition of the distinctive facial features-particularly when associated with markers of mitochondrial dysfunction and/or Irish ancestry-should suggest screening for the p.Trp22Arg NDUFB3 mutation to establish a genetic diagnosis, circumventing the requirement of muscle biopsy to direct genetic investigations.
孤立性复合体I缺乏是最常见的儿科线粒体疾病表现,预后不良且死亡率高。复合体I由44个结构亚基和至少10种辅助蛋白组成;其中29个亚基的突变迄今已与线粒体疾病相关,但基因型与表型的相关性有限,难以指导临床医生进行正确的基因诊断。
通过全外显子组测序、靶向捕获或候选基因测序对患者进行分析。对受影响个体进行临床表型分析。
我们从8个家庭中鉴定出一组10名患者(7个家庭为无血缘关系的爱尔兰血统),他们均身材矮小(<第9百分位数)且面部特征相似,包括前额突出、人中平滑和眼窝深陷,伴有复发性纯合子c.64T>C、p.Trp22Arg NDUFB3变异。两名同胞表现为原发性身材矮小,无明显代谢功能障碍。对三名患者的骨骼肌分析证实复合体I组装存在缺陷。
我们的报告强调,即使对于一些在急性代谢危机中出现且肌肉存在孤立性复合体I缺乏证据的患者,与p.Trp22Arg NDUFB3突变相关的长期预后也可能良好。识别独特的面部特征——尤其是与线粒体功能障碍标志物和/或爱尔兰血统相关时——应提示对p.Trp22Arg NDUFB3突变进行筛查以建立基因诊断,从而无需进行肌肉活检来指导基因研究。