Bendl Jaroslav, Musil Miloš, Štourač Jan, Zendulka Jaroslav, Damborský Jiří, Brezovský Jan
Loschmidt Laboratories, Department of Experimental Biology and Research Centre for Toxic Compounds in the Environment RECETOX, Masaryk University, Brno, Czech Republic.
Department of Information Systems, Faculty of Information Technology, Brno University of Technology, Brno, Czech Republic.
PLoS Comput Biol. 2016 May 25;12(5):e1004962. doi: 10.1371/journal.pcbi.1004962. eCollection 2016 May.
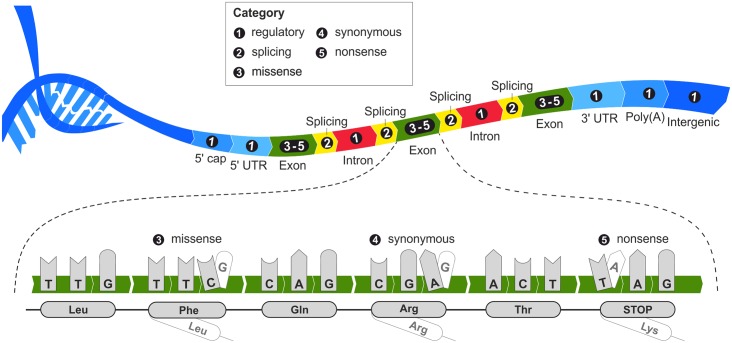
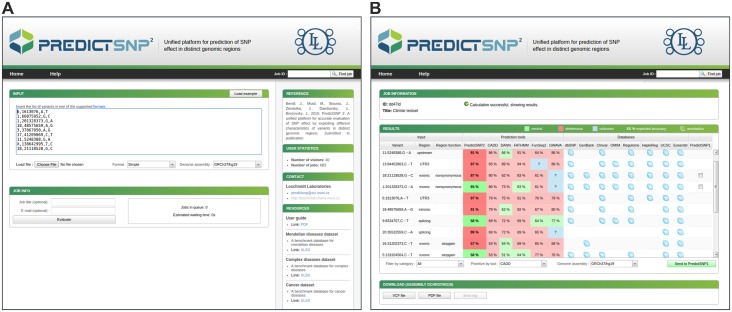
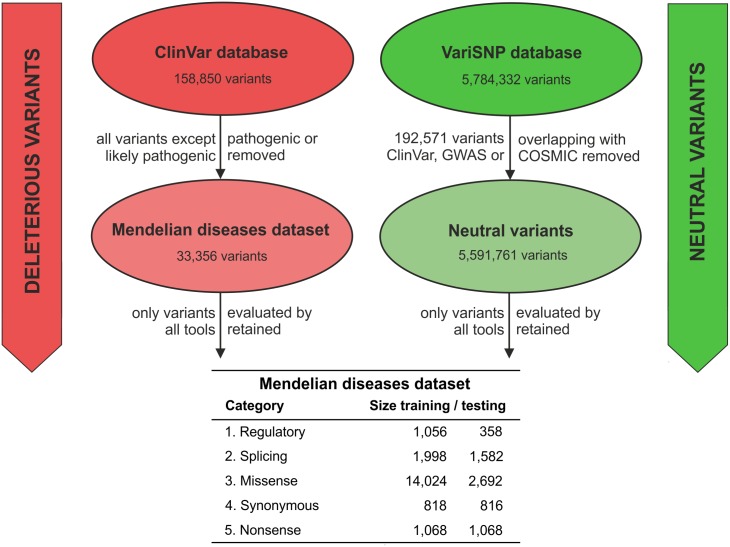
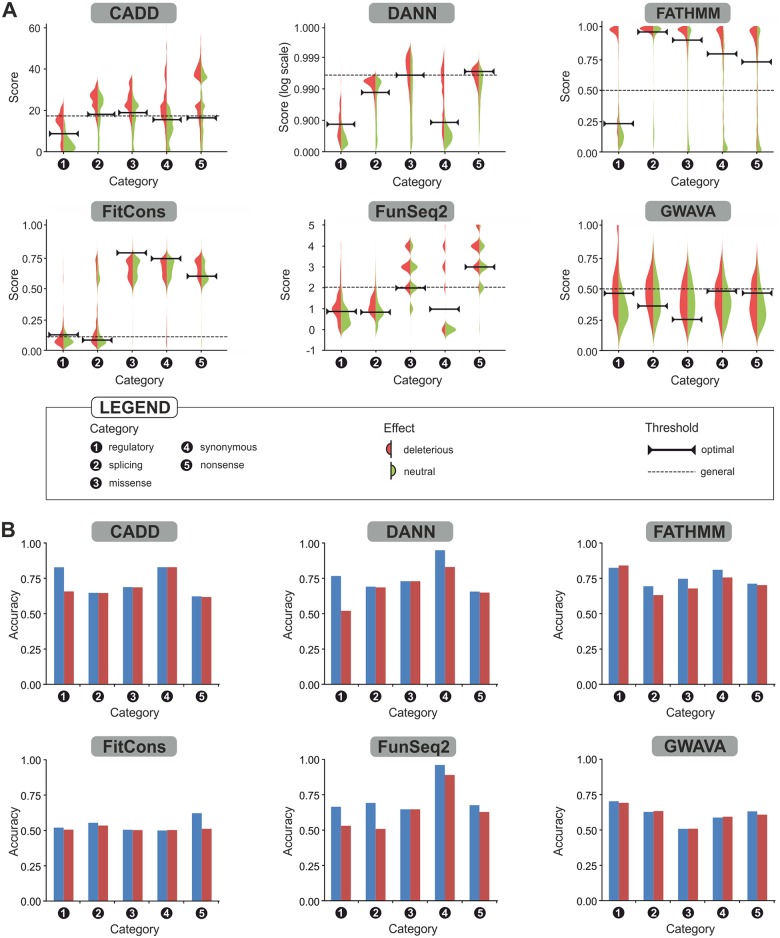
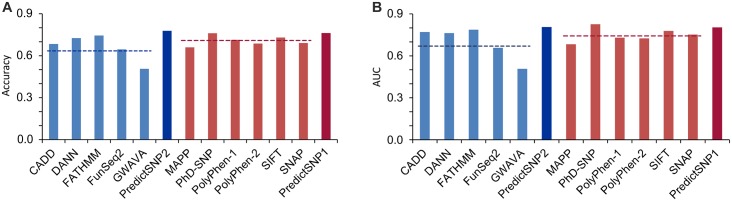
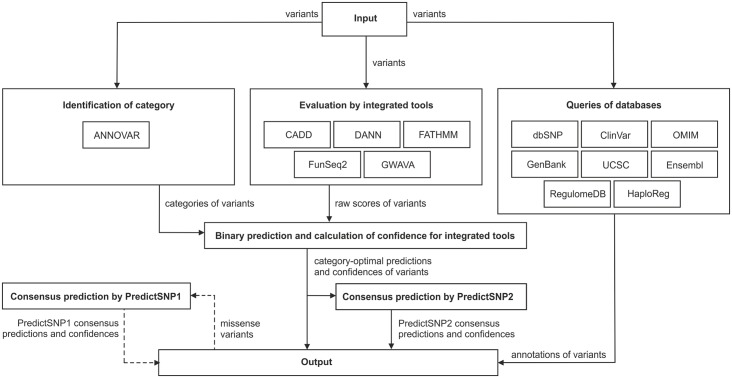
An important message taken from human genome sequencing projects is that the human population exhibits approximately 99.9% genetic similarity. Variations in the remaining parts of the genome determine our identity, trace our history and reveal our heritage. The precise delineation of phenotypically causal variants plays a key role in providing accurate personalized diagnosis, prognosis, and treatment of inherited diseases. Several computational methods for achieving such delineation have been reported recently. However, their ability to pinpoint potentially deleterious variants is limited by the fact that their mechanisms of prediction do not account for the existence of different categories of variants. Consequently, their output is biased towards the variant categories that are most strongly represented in the variant databases. Moreover, most such methods provide numeric scores but not binary predictions of the deleteriousness of variants or confidence scores that would be more easily understood by users. We have constructed three datasets covering different types of disease-related variants, which were divided across five categories: (i) regulatory, (ii) splicing, (iii) missense, (iv) synonymous, and (v) nonsense variants. These datasets were used to develop category-optimal decision thresholds and to evaluate six tools for variant prioritization: CADD, DANN, FATHMM, FitCons, FunSeq2 and GWAVA. This evaluation revealed some important advantages of the category-based approach. The results obtained with the five best-performing tools were then combined into a consensus score. Additional comparative analyses showed that in the case of missense variations, protein-based predictors perform better than DNA sequence-based predictors. A user-friendly web interface was developed that provides easy access to the five tools' predictions, and their consensus scores, in a user-understandable format tailored to the specific features of different categories of variations. To enable comprehensive evaluation of variants, the predictions are complemented with annotations from eight databases. The web server is freely available to the community at http://loschmidt.chemi.muni.cz/predictsnp2.
人类基因组测序项目得出的一个重要信息是,人类群体表现出约99.9%的基因相似性。基因组其余部分的变异决定了我们的身份、追溯我们的历史并揭示我们的遗传特征。对表型因果变异的精确界定在提供准确的个性化诊断、预后和遗传性疾病治疗方面起着关键作用。最近已经报道了几种用于实现这种界定的计算方法。然而,它们确定潜在有害变异的能力受到其预测机制未考虑不同类别变异存在这一事实的限制。因此,它们的输出偏向于变异数据库中最具代表性的变异类别。此外,大多数此类方法提供数值分数,而不是对变异有害性的二元预测或用户更容易理解的置信分数。我们构建了三个涵盖不同类型疾病相关变异的数据集,这些变异分为五类:(i) 调控变异,(ii) 剪接变异,(iii) 错义变异,(iv) 同义变异,以及 (v) 无义变异。这些数据集用于制定类别最优决策阈值,并评估六种变异优先级排序工具:CADD、DANN、FATHMM、FitCons、FunSeq2 和 GWAVA。该评估揭示了基于类别的方法的一些重要优势。然后将表现最佳的五个工具所获得的结果合并为一个共识分数。额外的比较分析表明,在错义变异的情况下,基于蛋白质的预测器比基于DNA序列的预测器表现更好。开发了一个用户友好的网络界面,以适合不同类别变异特定特征的用户可理解格式,提供对这五个工具的预测及其共识分数的便捷访问。为了能够对变异进行全面评估,预测结果辅以来自八个数据库的注释。该网络服务器可在http://loschmidt.chemi.muni.cz/predictsnp2上免费供社区使用。