Wall Jeffrey D, Stevison Laurie S
Institute for Human Genetics, University of California, San Francisco, California 94143
Institute for Human Genetics, University of California, San Francisco, California 94143 Department of Biological Sciences, Auburn University, Alabama 36849.
G3 (Bethesda). 2016 Aug 9;6(8):2265-71. doi: 10.1534/g3.116.029587.
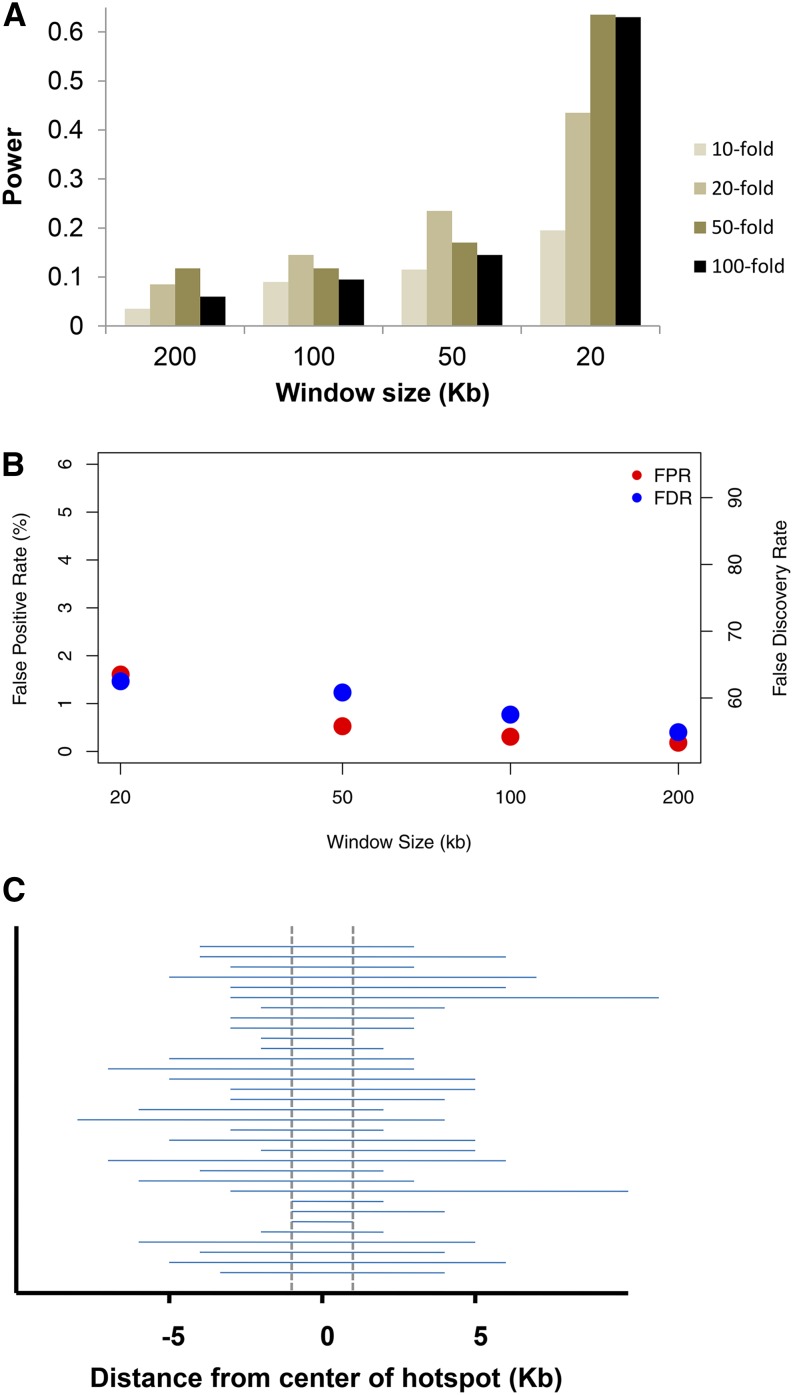
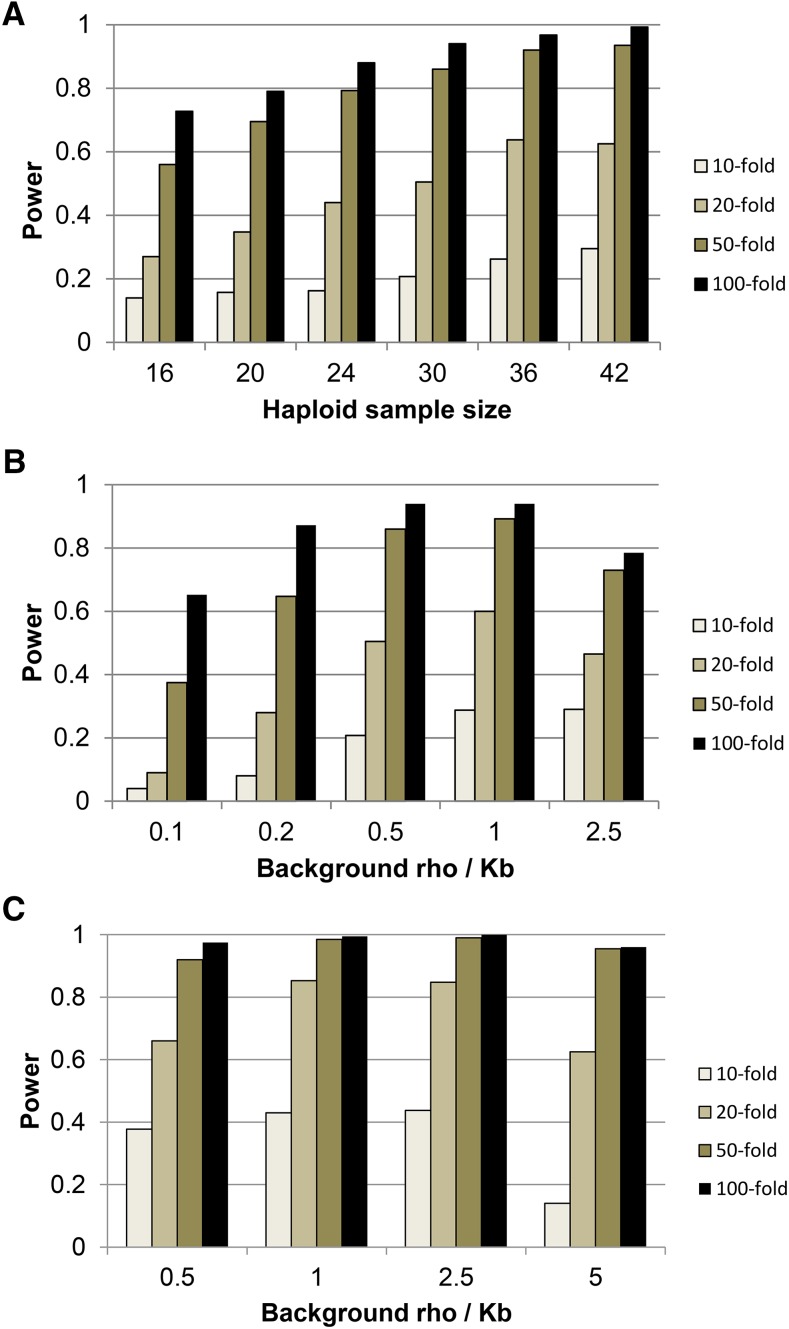
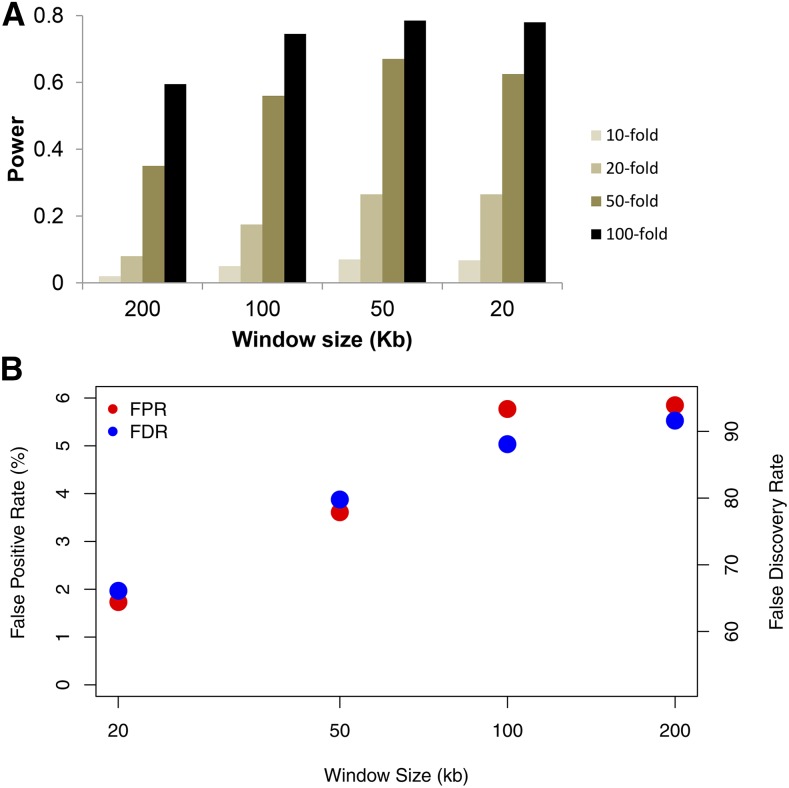
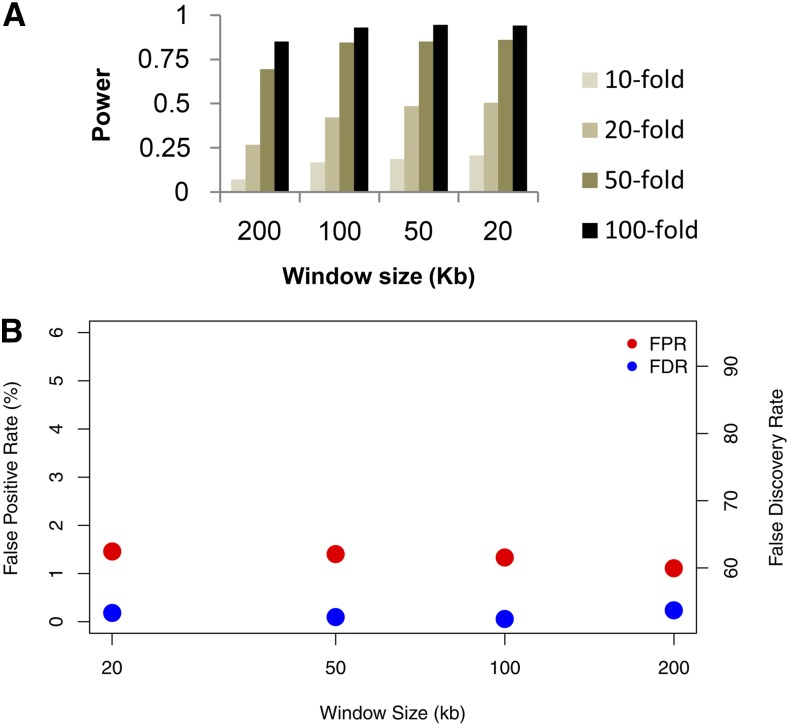
With recent advances in DNA sequencing technologies, it has become increasingly easy to use whole-genome sequencing of unrelated individuals to assay patterns of linkage disequilibrium (LD) across the genome. One type of analysis that is commonly performed is to estimate local recombination rates and identify recombination hotspots from patterns of LD. One method for detecting recombination hotspots, LDhot, has been used in a handful of species to further our understanding of the basic biology of recombination. For the most part, the effectiveness of this method (e.g., power and false positive rate) is unknown. In this study, we run extensive simulations to compare the effectiveness of three different implementations of LDhot. We find large differences in the power and false positive rates of these different approaches, as well as a strong sensitivity to the window size used (with smaller window sizes leading to more accurate estimation of hotspot locations). We also compared our LDhot simulation results with comparable simulation results obtained from a Bayesian maximum-likelihood approach for identifying hotspots. Surprisingly, we found that the latter computationally intensive approach had substantially lower power over the parameter values considered in our simulations.
随着DNA测序技术的最新进展,使用无关个体的全基因组测序来分析全基因组连锁不平衡(LD)模式变得越来越容易。一种常见的分析类型是估计局部重组率并从LD模式中识别重组热点。一种检测重组热点的方法LDhot已在少数物种中使用,以增进我们对重组基本生物学的理解。在很大程度上,这种方法的有效性(例如,功效和假阳性率)尚不清楚。在本研究中,我们进行了广泛的模拟,以比较LDhot的三种不同实现方式的有效性。我们发现这些不同方法在功效和假阳性率上存在很大差异,并且对所使用的窗口大小有很强的敏感性(较小的窗口大小导致对热点位置的估计更准确)。我们还将LDhot模拟结果与从用于识别热点的贝叶斯最大似然方法获得的可比模拟结果进行了比较。令人惊讶的是,我们发现后一种计算密集型方法在我们模拟中考虑的参数值范围内功效明显较低。