Li Xiaozhou, Tapmeyer Lukas, Bolte Michael, van de Streek Jacco
Department of Pharmacy, University of Copenhagen, Universitetsparken 2, DK-2100, Copenhagen, Denmark.
Institute for Inorganic and Analytical Chemistry, Goethe University, Max-von-Laue-Strasse 7, D-60438, Frankfurt am Main, Germany.
Chemphyschem. 2016 Aug 18;17(16):2496-502. doi: 10.1002/cphc.201600398. Epub 2016 Jun 8.
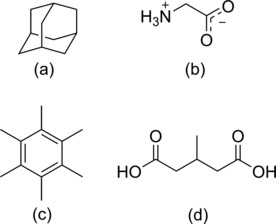
The excellent results of dispersion-corrected density functional theory (DFT-D) calculations for static systems have been well established over the past decade. The introduction of dynamics into DFT-D calculations is a target, especially for the field of molecular NMR crystallography. Four (13) C ss-NMR calibration compounds are investigated by single-crystal X-ray diffraction, molecular dynamics and DFT-D calculations. The crystal structure of 3-methylglutaric acid is reported. The rotator phases of adamantane and hexamethylbenzene at room temperature are successfully reproduced in the molecular dynamics simulations. The calculated (13) C chemical shifts of these compounds are in excellent agreement with experiment, with a root-mean-square deviation of 2.0 ppm. It is confirmed that a combination of classical molecular dynamics and DFT-D chemical shift calculation improves the accuracy of calculated chemical shifts.
过去十年间,色散校正密度泛函理论(DFT-D)对静态系统的出色计算结果已得到充分证实。将动力学引入DFT-D计算是一个目标,尤其在分子NMR晶体学领域。通过单晶X射线衍射、分子动力学和DFT-D计算对四种¹³C ss-NMR校准化合物进行了研究。报道了3-甲基戊二酸的晶体结构。在分子动力学模拟中成功再现了金刚烷和六甲基苯在室温下的旋转相。这些化合物的¹³C化学位移计算值与实验结果高度吻合,均方根偏差为2.0 ppm。证实了经典分子动力学和DFT-D化学位移计算相结合可提高化学位移计算的准确性。