Altrock Philipp M, Brendel Christian, Renella Raffaele, Orkin Stuart H, Williams David A, Michor Franziska
Department of Biostatistics and Computational Biology, Dana-Farber Cancer Institute, Boston, Massachusetts.
Department of Biostatistics, Harvard T.H. Chan School of Public Health, Boston, Massachuetts.
Am J Hematol. 2016 Sep;91(9):931-7. doi: 10.1002/ajh.24449. Epub 2016 Jul 14.
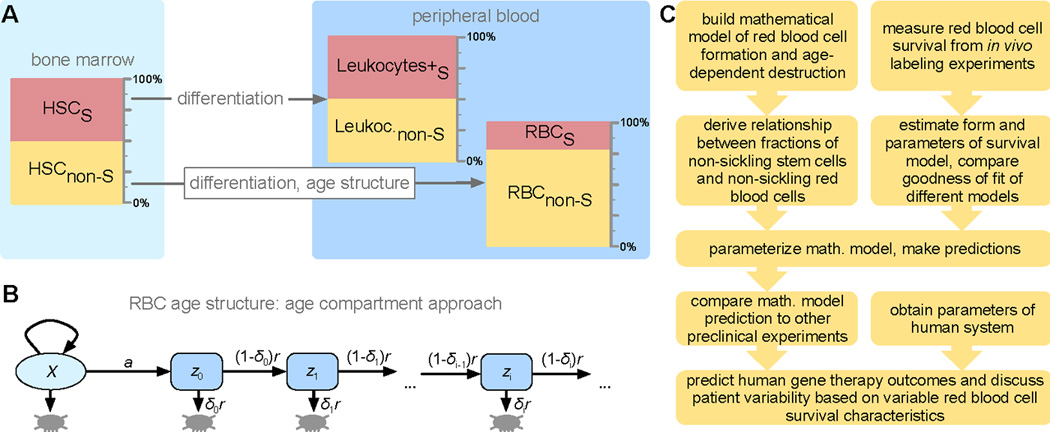
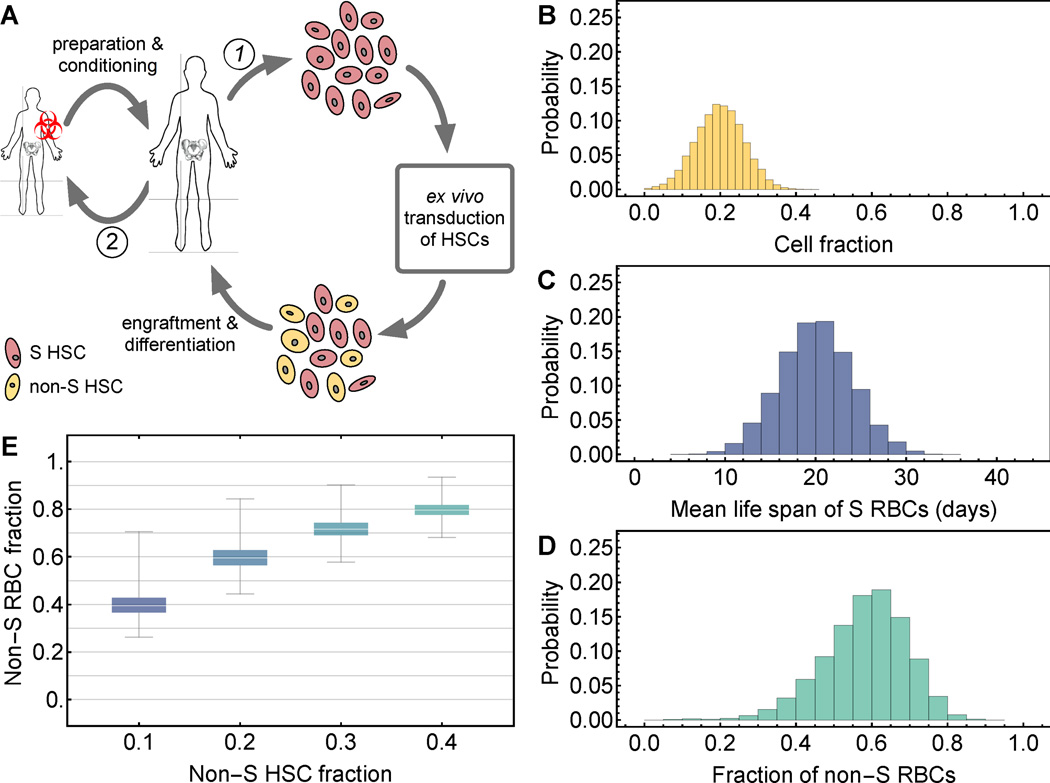
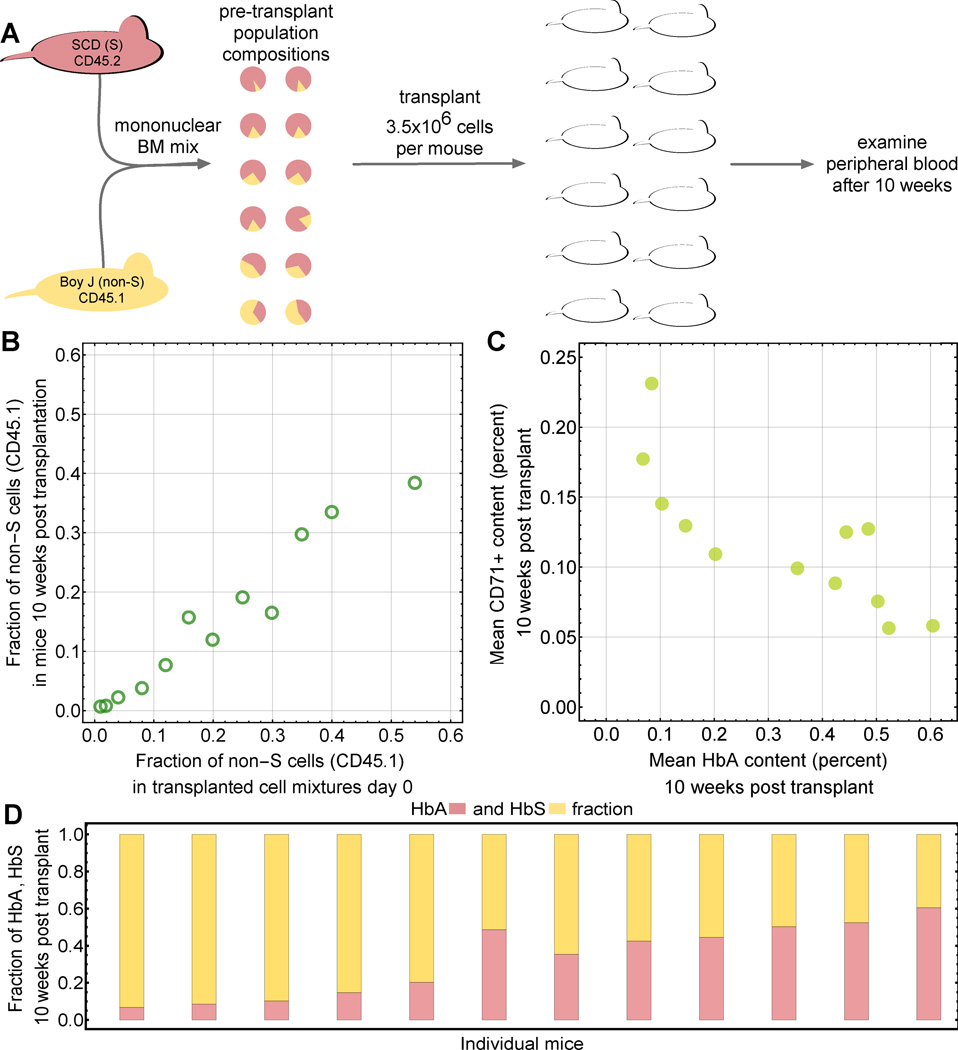
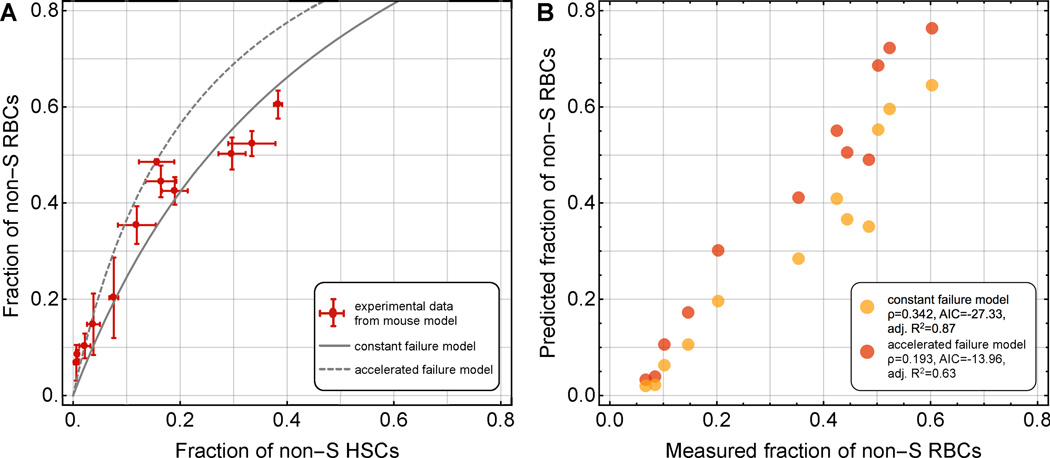
Recent advances in gene therapy and genome-engineering technologies offer the opportunity to correct sickle cell disease (SCD), a heritable disorder caused by a point mutation in the β-globin gene. The developmental switch from fetal γ-globin to adult β-globin is governed in part by the transcription factor (TF) BCL11A. This TF has been proposed as a therapeutic target for reactivation of γ-globin and concomitant reduction of β-sickle globin. In this and other approaches, genetic alteration of a portion of the hematopoietic stem cell (HSC) compartment leads to a mixture of sickling and corrected red blood cells (RBCs) in periphery. To reverse the sickling phenotype, a certain proportion of corrected RBCs is necessary; the degree of HSC alteration required to achieve a desired fraction of corrected RBCs remains unknown. To address this issue, we developed a mathematical model describing aging and survival of sickle-susceptible and normal RBCs; the former can have a selective survival advantage leading to their overrepresentation. We identified the level of bone marrow chimerism required for successful stem cell-based gene therapies in SCD. Our findings were further informed using an experimental mouse model, where we transplanted mixtures of Berkeley SCD and normal murine bone marrow cells to establish chimeric grafts in murine hosts. Our integrative theoretical and experimental approach identifies the target frequency of HSC alterations required for effective treatment of sickling syndromes in humans. Our work replaces episodic observations of such target frequencies with a mathematical modeling framework that covers a large and continuous spectrum of chimerism conditions. Am. J. Hematol. 91:931-937, 2016. © 2016 Wiley Periodicals, Inc.
基因治疗和基因组工程技术的最新进展为纠正镰状细胞病(SCD)提供了机会,SCD是一种由β-珠蛋白基因突变引起的遗传性疾病。从胎儿γ-珠蛋白到成人β-珠蛋白的发育转换部分受转录因子(TF)BCL11A调控。该转录因子已被提议作为重新激活γ-珠蛋白并同时减少β-镰状珠蛋白的治疗靶点。在这种及其他方法中,部分造血干细胞(HSC)区室的基因改变会导致外周血中镰状红细胞和纠正后的红细胞(RBC)混合存在。为了逆转镰状表型,需要一定比例的纠正后RBC;实现所需比例的纠正后RBC所需的HSC改变程度尚不清楚。为了解决这个问题,我们开发了一个数学模型来描述镰状易感性和正常RBC的衰老和存活情况;前者可能具有选择性存活优势,导致其数量过多。我们确定了SCD中基于干细胞的基因治疗成功所需的骨髓嵌合水平。我们使用实验小鼠模型进一步验证了我们的发现,在该模型中,我们移植了伯克利SCD小鼠和正常小鼠的骨髓细胞混合物,以在小鼠宿主中建立嵌合移植物。我们综合的理论和实验方法确定了有效治疗人类镰状综合征所需的HSC改变的目标频率。我们的工作用一个涵盖广泛且连续的嵌合情况谱的数学建模框架取代了对这种目标频率的偶发性观察。《美国血液学杂志》91:931 - 937,2016年。© 2016威利期刊公司。