Department of Pathology, Beth Israel Deaconess Medical Center BIDMC East/Dana 615, 330 Brookline Avenue, Boston, Massachusetts 02215, USA.
Department of Systems Biology, Harvard Medical School, Boston, Massachusetts 02215, USA.
Nat Commun. 2016 Jun 15;7:11881. doi: 10.1038/ncomms11881.
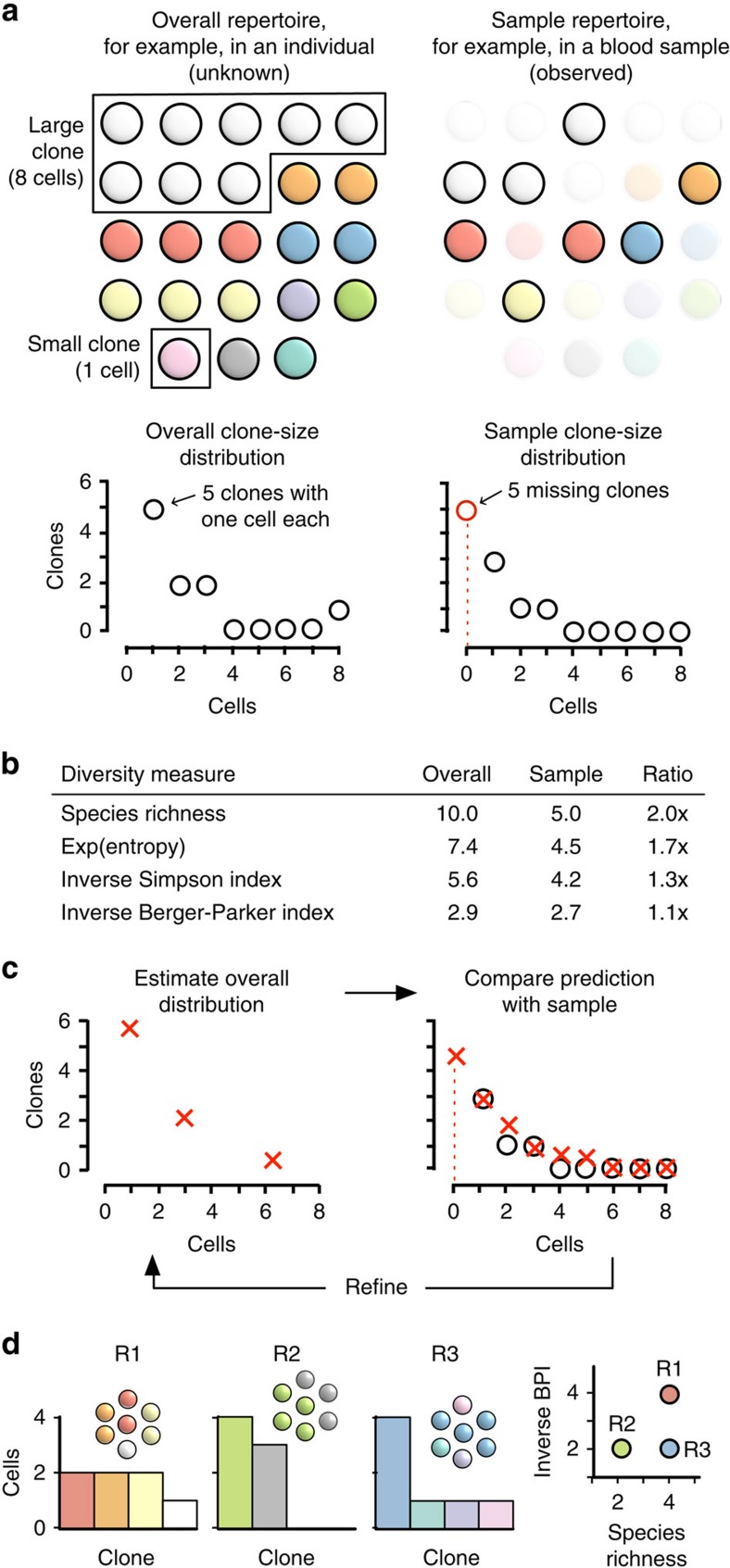
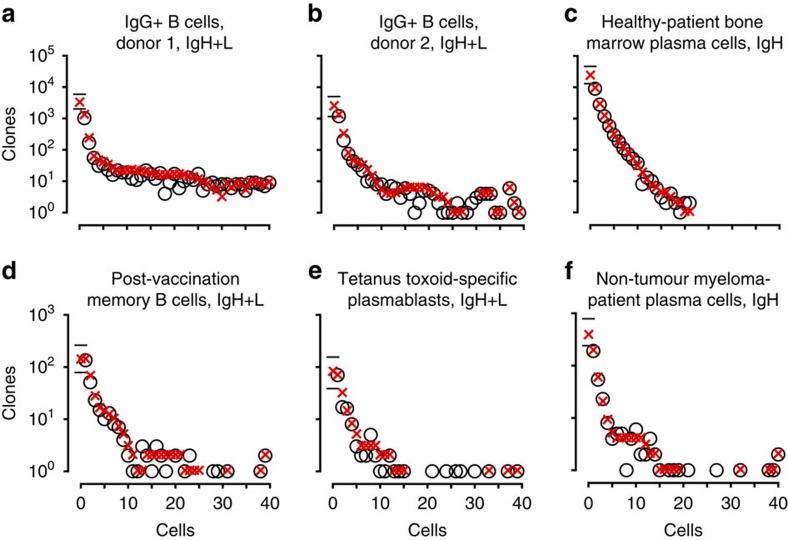
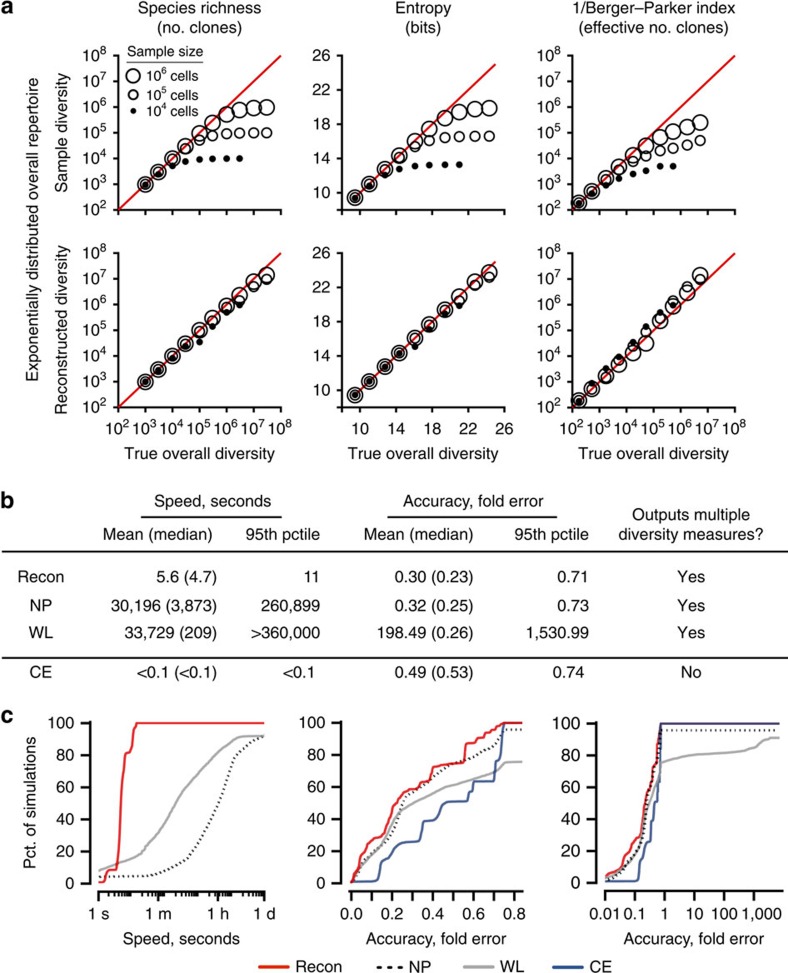
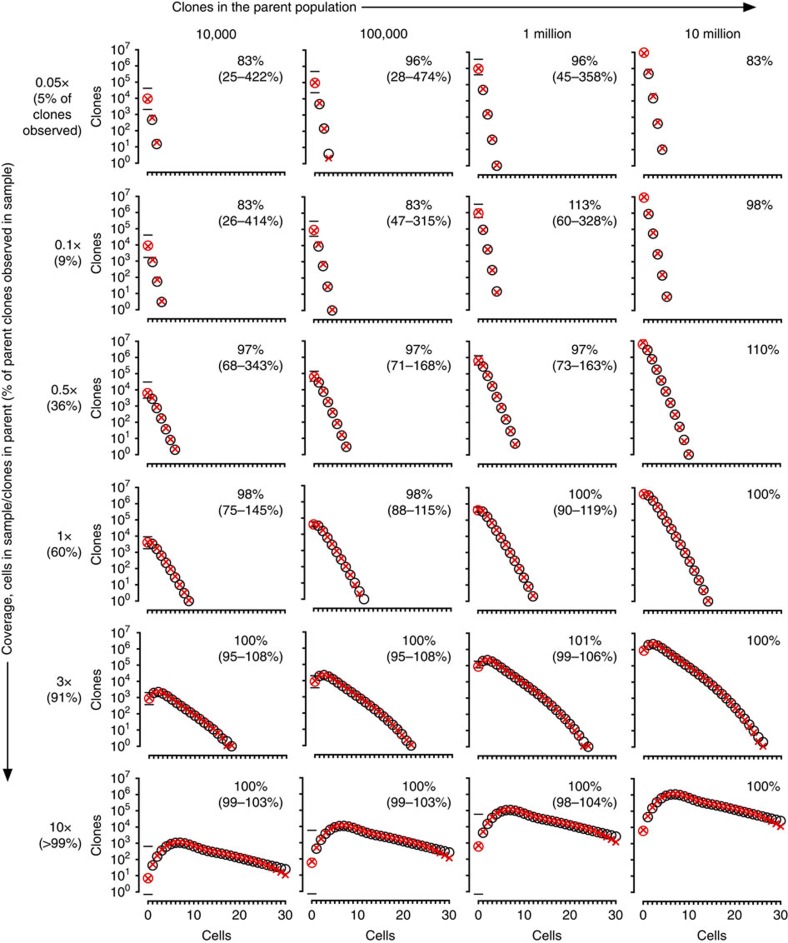
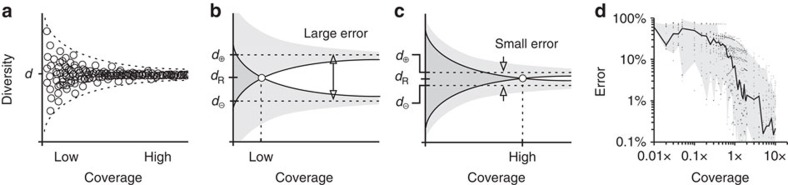
The diversity of an organism's B- and T-cell repertoires is both clinically important and a key measure of immunological complexity. However, diversity is hard to estimate by current methods, because of inherent uncertainty in the number of B- and T-cell clones that will be missing from a blood or tissue sample by chance (the missing-species problem), inevitable sampling bias, and experimental noise. To solve this problem, we developed Recon, a modified maximum-likelihood method that outputs the overall diversity of a repertoire from measurements on a sample. Recon outputs accurate, robust estimates by any of a vast set of complementary diversity measures, including species richness and entropy, at fractional repertoire coverage. It also outputs error bars and power tables, allowing robust comparisons of diversity between individuals and over time. We apply Recon to in silico and experimental immune-repertoire sequencing data sets as proof of principle for measuring diversity in large, complex systems.
生物体 B 细胞和 T 细胞受体库的多样性具有重要的临床意义,也是衡量免疫复杂性的关键指标。然而,由于血液或组织样本中偶然缺失 B 细胞和 T 细胞克隆的数量存在固有不确定性(物种缺失问题)、不可避免的采样偏差和实验噪声,当前的方法很难估计多样性。为了解决这个问题,我们开发了 Recon,这是一种改进的最大似然法,可从样本测量中输出受体库的整体多样性。Recon 通过大量互补多样性测量中的任何一种(包括物种丰富度和熵),在受体库部分覆盖的情况下输出准确、稳健的估计值。它还输出误差条和功效表,允许在个体之间和随时间进行稳健的多样性比较。我们将 Recon 应用于免疫受体库测序的计算机模拟和实验数据集,以证明在大型复杂系统中测量多样性的原理。