Goker-Alpan Ozlem, Longo Nicola, McDonald Marie, Shankar Suma P, Schiffmann Raphael, Chang Peter, Shen Yinghua, Pano Arian
Lysosomal Disorders Unit, Fairfax, VA, USA.
University of Utah, Salt Lake City, UT, USA.
Drug Des Devel Ther. 2016 May 25;10:1771-81. doi: 10.2147/DDDT.S102761. eCollection 2016.
Following a drug manufacturing process change, safety/efficacy of agalsidase alfa were evaluated in enzyme replacement therapy (ERT)-naïve children with Fabry disease.
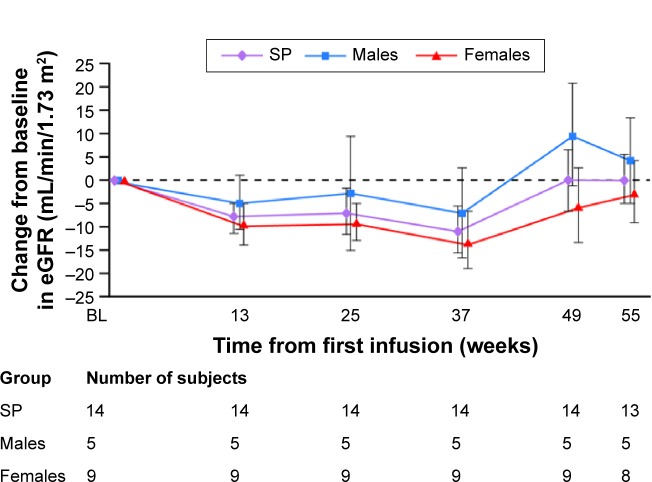
In an open-label, multicenter, Phase II study (HGT-REP-084; Shire), 14 children aged ≥7 years received 0.2 mg/kg agalsidase alfa every other week for 55 weeks. Primary endpoints: safety, changes in autonomic function (2-hour Holter monitoring). Secondary endpoints: estimated glomerular filtration rate, left ventricular mass index (LVMI), midwall fractional shortening, pharmacodynamic parameters, and patient-reported quality-of-life.
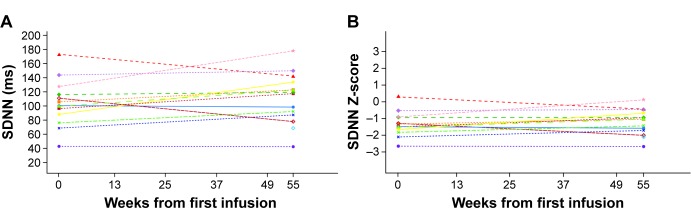
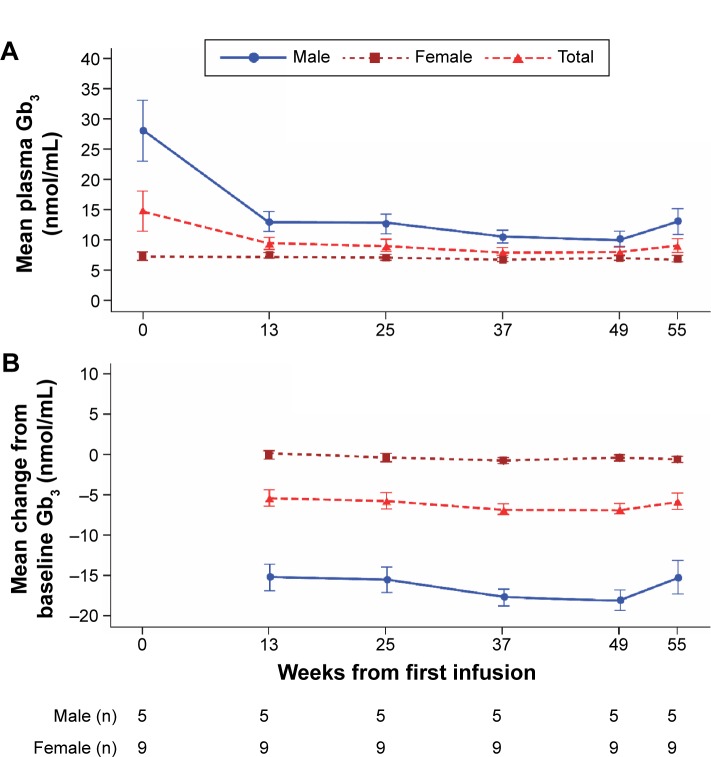
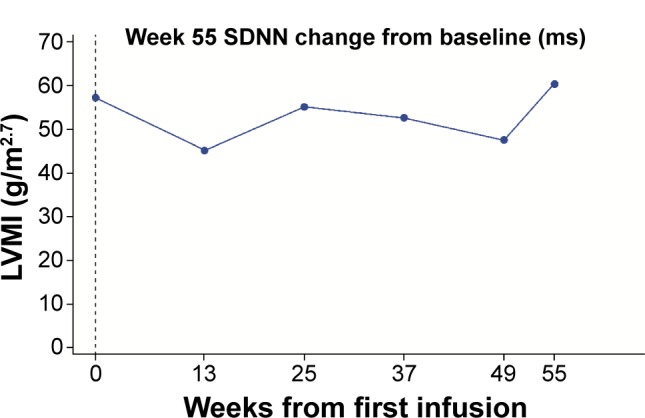
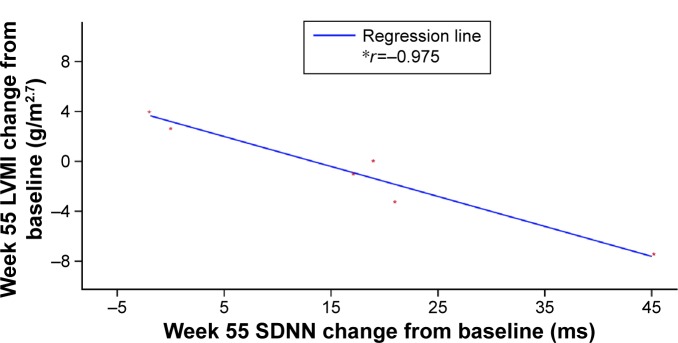
Among five boys (median 10.2 [range 6.7, 14.4] years) and nine girls (14.8 [10.1, 15.9] years), eight patients experienced infusion-related adverse events (vomiting, n=4; nausea, n=3; dyspnea, n=3; chest discomfort, n=2; chills, n=2; dizziness, n=2; headache, n=2). One of these had several hypersensitivity episodes. However, no patient discontinued for safety reasons and no serious adverse events occurred. One boy developed immunoglobulin G (IgG) and neutralizing antidrug antibodies. Overall, no deterioration in cardiac function was observed in seven patients with low/abnormal SDNN (standard deviation of all filtered RR intervals; <100 ms) and no left ventricular hypertrophy: mean (SD) baseline SDNN, 81.6 (20.9) ms; mean (95% confidence interval [CI]) change from baseline to week 55, 17.4 (2.9, 31.9) ms. Changes in SDNN correlated with changes in LVMI (r=-0.975). No change occurred in secondary efficacy endpoints: mean (95% CI) change from baseline at week 55 in LVMI, 0.16 (-3.3, 3.7) g/m(2.7); midwall fractional shortening, -0.62% (-2.7%, 1.5%); estimated glomerular filtration rate, 0.15 (-11.4, 11.7) mL/min/1.73 m(2); urine protein, -1.8 (-6.0, 2.4) mg/dL; urine microalbumin, 0.6 (-0.5, 1.7) mg/dL; plasma globotriaosylceramide (Gb3), -5.71 (-10.8, -0.6) nmol/mL; urinary Gb3, -1,403.3 (-3,714.0, 907.4) nmol/g creatinine, or clinical quality-of-life outcomes.
Fifty-five weeks' agalsidase alfa ERT at 0.2 mg/kg every other week was well tolerated. Disease progression may be slowed when ERT is started prior to major organ dysfunction.
https://ClinicalTrials.gov identifier NCT01363492.
在药物生产工艺变更后,对初治法布里病儿童进行阿加糖酶α酶替代疗法(ERT)的安全性/有效性进行了评估。
在一项开放标签、多中心的II期研究(HGT-REP-084;夏尔公司)中,14名年龄≥7岁的儿童每隔一周接受0.2mg/kg阿加糖酶α治疗,持续55周。主要终点:安全性、自主神经功能变化(24小时动态心电图监测)。次要终点:估计肾小球滤过率、左心室质量指数(LVMI)、室壁中层缩短率、药效学参数以及患者报告的生活质量。
在5名男孩(中位年龄10.2[范围6.7,14.4]岁)和9名女孩(14.8[10.1,15.9]岁)中,8名患者发生了输液相关不良事件(呕吐,n = 4;恶心,n = 3;呼吸困难,n = 3;胸部不适,n = 2;寒战,n = 2;头晕,n = 2;头痛,n = 2)。其中1例发生了多次过敏反应。然而,没有患者因安全原因停药,也未发生严重不良事件。1名男孩产生了免疫球蛋白G(IgG)和中和性抗药物抗体。总体而言,7名SDNN(所有滤波后RR间期的标准差;<100ms)低/异常且无左心室肥厚的患者未观察到心脏功能恶化:平均(SD)基线SDNN为81.6(20.9)ms;从基线到第55周的平均(95%置信区间[CI])变化为17.4(2.9,31.9)ms。SDNN的变化与LVMI的变化相关(r = -0.975)。次要疗效终点无变化:第55周时LVMI相对于基线的平均(95%CI)变化为0.16(-3.3,3.7)g/m².⁷;室壁中层缩短率为-0.62%(-2.7%,1.5%);估计肾小球滤过率为0.15(-11.4,11.7)mL/min/1.73m²;尿蛋白为-1.8(-6.0,2.4)mg/dL;尿微量白蛋白为0.6(-0.5,1.7)mg/dL;血浆球三糖神经酰胺(Gb3)为-5.71(-10.8, -0.6)nmol/mL;尿Gb3为-1403.3(-3714.0,907.4)nmol/g肌酐,或临床生活质量结果。
每隔一周给予0.2mg/kg阿加糖酶α进行55周的ERT耐受性良好。在主要器官功能障碍之前开始ERT可能会减缓疾病进展。