Mahil Satveer K, Twelves Sophie, Farkas Katalin, Setta-Kaffetzi Niovi, Burden A David, Gach Joanna E, Irvine Alan D, Képíró László, Mockenhaupt Maja, Oon Hazel H, Pinner Jason, Ranki Annamari, Seyger Marieke M B, Soler-Palacin Pere, Storan Eoin R, Tan Eugene S, Valeyrie-Allanore Laurence, Young Helen S, Trembath Richard C, Choon Siew-Eng, Szell Marta, Bata-Csorgo Zsuzsanna, Smith Catherine H, Di Meglio Paola, Barker Jonathan N, Capon Francesca
Division of Genetics and Molecular Medicine, King's College London, London, UK.
MTA-SZTE Dermatological Research Group, Szeged, Hungary.
J Invest Dermatol. 2016 Nov;136(11):2251-2259. doi: 10.1016/j.jid.2016.06.618. Epub 2016 Jul 5.
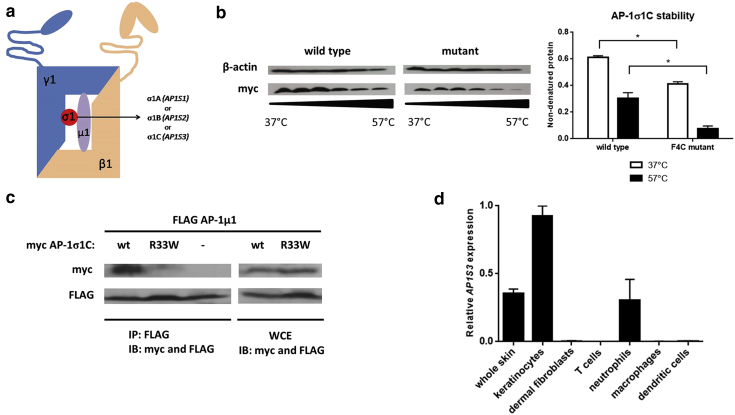
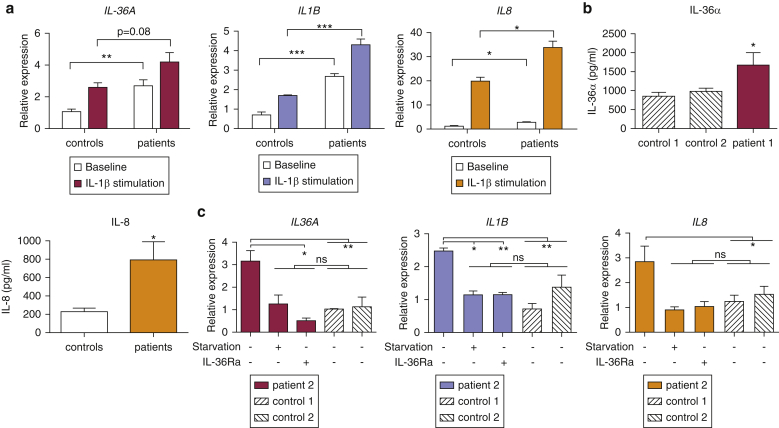
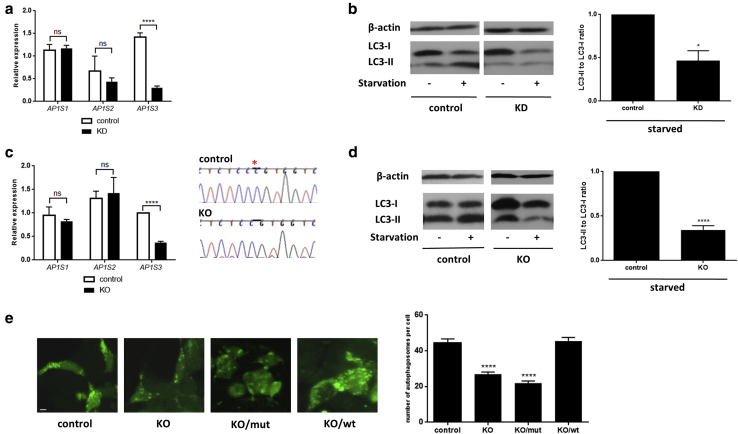
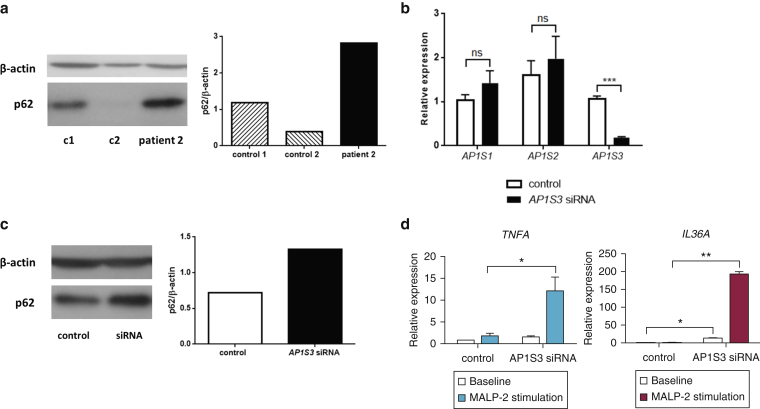
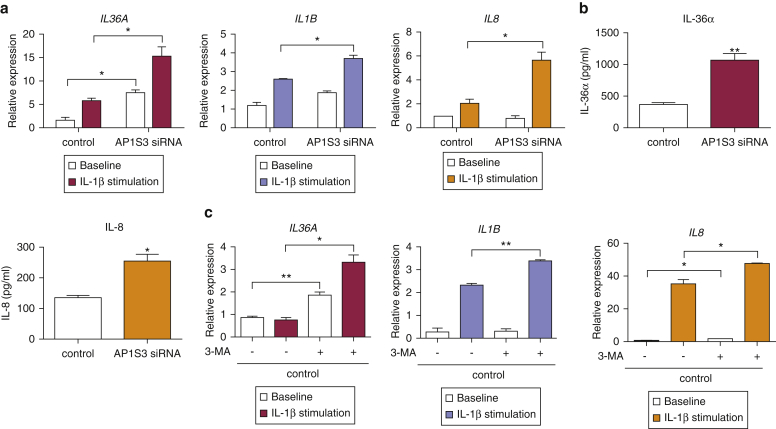
Prominent skin involvement is a defining characteristic of autoinflammatory disorders caused by abnormal IL-1 signaling. However, the pathways and cell types that drive cutaneous autoinflammatory features remain poorly understood. We sought to address this issue by investigating the pathogenesis of pustular psoriasis, a model of autoinflammatory disorders with predominant cutaneous manifestations. We specifically characterized the impact of mutations affecting AP1S3, a disease gene previously identified by our group and validated here in a newly ascertained patient resource. We first showed that AP1S3 expression is distinctively elevated in keratinocytes. Because AP1S3 encodes a protein implicated in autophagosome formation, we next investigated the effects of gene silencing on this pathway. We found that AP1S3 knockout disrupts keratinocyte autophagy, causing abnormal accumulation of p62, an adaptor protein mediating NF-κB activation. We showed that as a consequence, AP1S3-deficient cells up-regulate IL-1 signaling and overexpress IL-36α, a cytokine that is emerging as an important mediator of skin inflammation. These abnormal immune profiles were recapitulated by pharmacological inhibition of autophagy and verified in patient keratinocytes, where they were reversed by IL-36 blockade. These findings show that keratinocytes play a key role in skin autoinflammation and identify autophagy modulation of IL-36 signaling as a therapeutic target.
显著的皮肤受累是由异常白细胞介素-1信号传导引起的自身炎症性疾病的一个决定性特征。然而,驱动皮肤自身炎症特征的途径和细胞类型仍知之甚少。我们试图通过研究脓疱型银屑病的发病机制来解决这个问题,脓疱型银屑病是一种以皮肤表现为主的自身炎症性疾病模型。我们特别研究了影响AP1S3突变的影响,AP1S3是我们团队先前鉴定的一个疾病基因,并在新确定的患者资源中得到验证。我们首先表明,AP1S3在角质形成细胞中的表达明显升高。由于AP1S3编码一种与自噬体形成有关的蛋白质,我们接下来研究了基因沉默对该途径的影响。我们发现,AP1S3基因敲除会破坏角质形成细胞的自噬,导致p62异常积累,p62是一种介导NF-κB激活的衔接蛋白。我们表明,结果是,缺乏AP1S3的细胞上调白细胞介素-1信号传导并过度表达白细胞介素-36α,白细胞介素-36α是一种正在成为皮肤炎症重要介质的细胞因子。通过自噬的药理学抑制重现了这些异常免疫特征,并在患者角质形成细胞中得到验证,在这些细胞中,白细胞介素-36阻断可逆转这些特征。这些发现表明角质形成细胞在皮肤自身炎症中起关键作用,并确定白细胞介素-36信号传导的自噬调节作为一个治疗靶点。