Sheth Jayesh, Datar Chaitanya, Mistri Mehul, Bhavsar Riddhi, Sheth Frenny, Shah Krati
Department of Biochemical and Molecular Genetics, FRIGE's Institute of Human Genetics, FRIGE House, Jodhpur Gam Road, Satellite, Ahmedabad, 380015, Gujarat, India.
Sahyadari Medical Genetics and Tissue engineering facility (SMGTEF), Pune, 411005, India.
BMC Pediatr. 2016 Jul 11;16:88. doi: 10.1186/s12887-016-0626-6.
GM2 gangliosidosis-AB variants a rare autosomal recessive neurodegenerative disorder occurring due to deficiency of GM2 activator protein resulting from the mutation in GM2A gene. Only seven mutations in nine cases have been reported from different population except India.
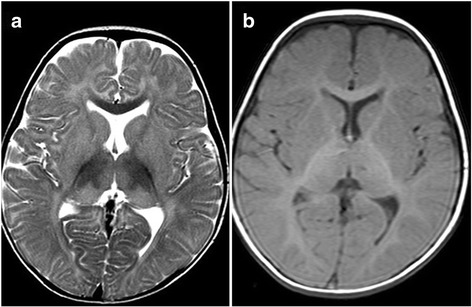
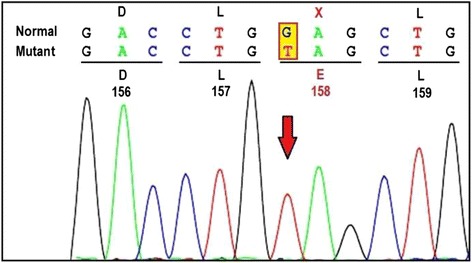
Present case is a one year old male born to 3rd degree consanguineous Indian parents from Maharashtra. He was presented with global developmental delay, hypotonia and sensitive to hyperacusis. Horizontal nystagmus and cherry red spot was detected during ophthalmic examination. MRI of brain revealed putaminal hyperintensity and thalamic hypointensity with some unmyelinated white matter in T2/T1 weighted images. Initially he was suspected having Tay-Sachs disease and finally diagnosed as GM2 gangliosidosis, AB variant due to truncated protein caused by nonsense mutation c.472 G > T (p.E158X) in GM2Agene.
Children with phenotypic presentation as GM2 gangliosidosis (Tay-Sachs or Sandhoff disease) and normal enzyme activity of β-hexosaminidase-A and -B in leucocytes need to be investigated for GM2 activator protein deficiency.
GM2神经节苷脂贮积症AB变异型是一种罕见的常染色体隐性神经退行性疾病,由于GM2A基因突变导致GM2激活蛋白缺乏所致。除印度外,不同人群中仅报道了9例中的7种突变。
本病例为一名1岁男性,其父母为来自马哈拉施特拉邦的三级近亲结婚的印度人。他出现全面发育迟缓、肌张力减退和对听觉过敏敏感。眼科检查发现水平性眼球震颤和樱桃红斑。脑部MRI显示在T2/T1加权图像中壳核高信号和丘脑低信号,伴有一些未髓鞘化的白质。最初怀疑他患有泰-萨克斯病,最终诊断为GM2神经节苷脂贮积症AB变异型,是由于GM2A基因中无义突变c.472 G>T(p.E158X)导致的截短蛋白所致。
对于表现为GM2神经节苷脂贮积症(泰-萨克斯病或桑德霍夫病)且白细胞中β-己糖胺酶A和B酶活性正常的儿童,需要调查是否存在GM2激活蛋白缺乏。