Martins Carla, Brunel-Guitton Catherine, Lortie Anne, Gauvin France, Morales Carlos R, Mitchell Grant A, Pshezhetsky Alexey V
CHU Ste-Justine, University of Montreal, Montreal, QC, Canada.
Department of Anatomy and Cell Biology, McGill University, Montreal, QC, Canada.
Mol Genet Metab Rep. 2017 Apr 7;11:24-29. doi: 10.1016/j.ymgmr.2017.01.017. eCollection 2017 Jun.
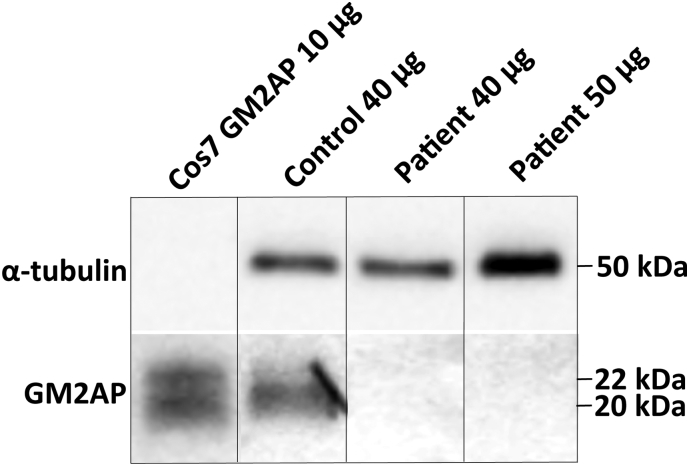
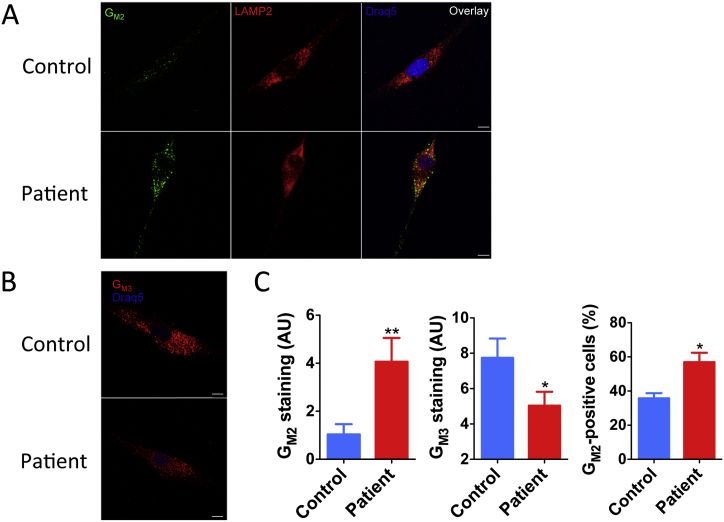
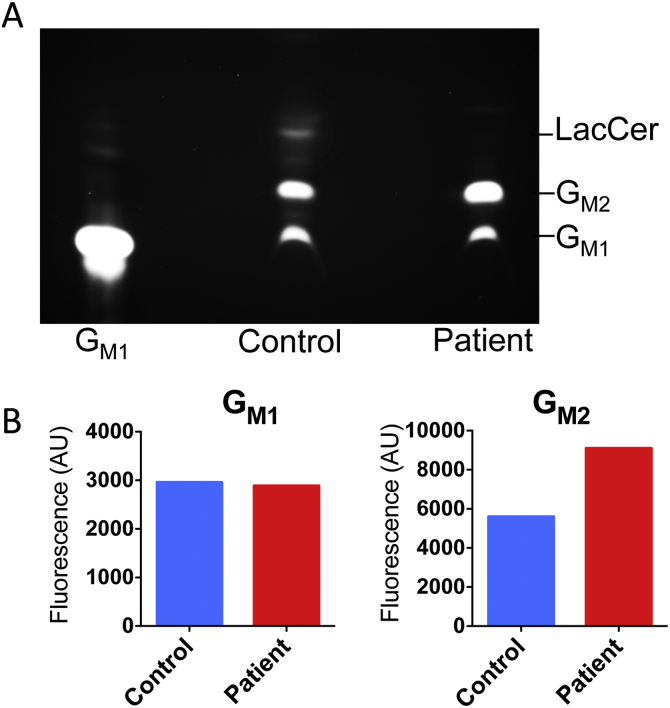
G-gangliosidosis, AB variant is an extremely rare autosomal recessive inherited disorder caused by mutations in the gene that encodes G ganglioside activator protein (GM2AP). GM2AP is necessary for solubilisation of G ganglioside in endolysosomes and its presentation to β-hexosaminidase A. Conversely GM2AP deficiency impairs lysosomal catabolism of G ganglioside, leading to its storage in cells and tissues. We describe a 9-year-old child with an unusual juvenile clinical onset of G-gangliosidosis AB. At the age of 3 years he presented with global developmental delay, progressive epilepsy, intellectual disability, axial hypertonia, spasticity, seizures and ataxia, but without the macular cherry-red spots typical for G gangliosidosis. Brain MRI detected a rapid onset of diffuse atrophy, whereas whole exome sequencing showed that the patient is a compound heterozygote for two mutations in : a novel nonsense mutation, c.259G > T (p.E87X) and a missense mutation c.164C > T (p.P55L) that was recently identified in homozygosity in patients of a Saudi family with a progressive chorea-dementia syndrome. Western blot analysis showed an absence of GM2AP in cultured fibroblasts from the patient, suggesting that both mutations interfere with the synthesis and/or folding of the protein. Finally, impaired catabolism of G ganglioside in the patient's fibroblasts was demonstrated by metabolic labeling with fluorescently labeled G ganglioside and by immunohistochemistry with anti-G and anti-G antibodies. Our observation expands the molecular and clinical spectrum of molecular defects linked to G-gangliosidosis and suggests novel diagnostic approach by whole exome sequencing and perhaps ganglioside analysis in cultured patient's cells.
G-神经节苷脂沉积症AB变异型是一种极其罕见的常染色体隐性遗传性疾病,由编码G神经节苷脂激活蛋白(GM2AP)的基因突变引起。GM2AP对于神经节苷脂在溶酶体内的溶解及其向β-己糖胺酶A的呈递是必需的。相反,GM2AP缺乏会损害神经节苷脂的溶酶体分解代谢,导致其在细胞和组织中蓄积。我们描述了一名9岁儿童,其患有不寻常的青少年期临床起病的G-神经节苷脂沉积症AB变异型。3岁时,他出现全面发育迟缓、进行性癫痫、智力残疾、轴性张力亢进、痉挛、癫痫发作和共济失调,但没有G-神经节苷脂沉积症典型的黄斑樱桃红斑。脑部磁共振成像检测到弥漫性萎缩迅速出现,而全外显子组测序显示该患者是两个突变的复合杂合子:一个新的无义突变c.259G>T(p.E87X)和一个错义突变c.164C>T(p.P55L),后者最近在一个患有进行性舞蹈症-痴呆综合征的沙特家族患者中以纯合子形式被发现。蛋白质印迹分析显示患者培养的成纤维细胞中不存在GM2AP,提示这两个突变均干扰了该蛋白的合成和/或折叠。最后,通过用荧光标记的神经节苷脂进行代谢标记以及用抗G和抗G抗体进行免疫组织化学,证实了患者成纤维细胞中神经节苷脂的分解代谢受损。我们的观察扩展了与G-神经节苷脂沉积症相关的分子缺陷的分子和临床谱,并提示通过全外显子组测序以及可能对患者培养细胞进行神经节苷脂分析的新诊断方法。