Zhao Guolian, Zhang Xun, Wang Changmin, Wang Guoqing, Li Fan
Department of Pathogenobiology, The Key Laboratory of Zoonosis, Chinese Ministry of Education, Norman Bethune College of Basic Medicine, Jilin University, Changchun, Jilin, 130021, China.
Virol J. 2016 Jul 28;13:130. doi: 10.1186/s12985-016-0578-3.
Coxsackievirus A16 (CV-A16), a major etiopathologic cause of pediatric hand, foot, and mouth disease (HFMD) worldwide, has been reported to have caused several fatalities. Revealing the evolutionary and epidemiologic dynamics of CV-A16 across time and space is central to understanding its outbreak potential.
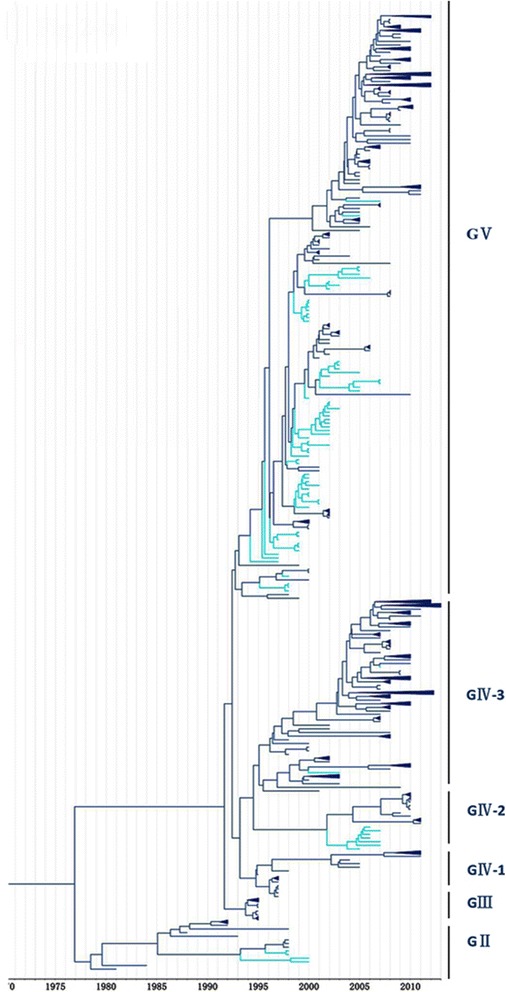
In this study, we isolated six CV-A16 strains in China's Jilin province and construct a maximum clade credibility (MCC) tree for CV-A16 VP1 gene by the Bayesian Markov Chain Monte Carlo method using 708 strains from GenBank with epidemiological information. The evolution characteristics of CV-A16 VP1 gene was also analysed dynamicly through Bayesian skyline plot.
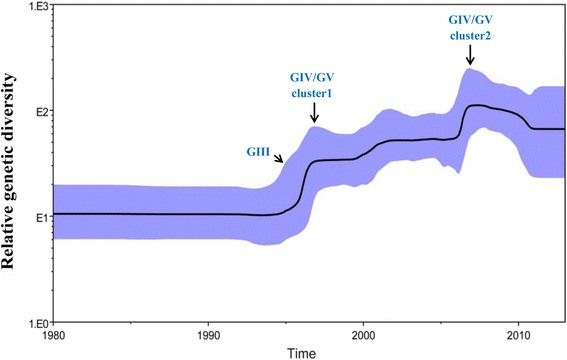
All CV-A16 strains identified could be classified into five major genogroups, denoted by GI-GV. GIV and GV have co-circulated in China since 2007, and the CV-A16 epidemic strain isolated in the Jilin province, China, can be classified as GIV-3. The CV-A16 genogroups circulating recently in China have the same ancestor since 2007. The genetic diversity of the CV-A16 VP1 gene shows a continuous increase since the mid-1990s, with sharp increases in genetic diversity in 1997 and 2007 and reached peak in 2007. Very low genetic diversity existed after 2010. The CV-A16 VP1 gene evolutionary rate was 6.656E-3 substitutions per site per year.
We predicted the dynamic phylogenetic trends, which indicate outbreak trends of CV-A16, and provide theoretical foundations for clinical prevention and treatment of HFMD which caused by a CV-A16.
柯萨奇病毒A16(CV-A16)是全球小儿手足口病(HFMD)的主要病因,据报道已导致数人死亡。揭示CV-A16在时间和空间上的进化及流行病学动态对于了解其爆发潜力至关重要。
在本研究中,我们在中国吉林省分离出6株CV-A16毒株,并使用来自GenBank的708株带有流行病学信息的毒株,通过贝叶斯马尔可夫链蒙特卡罗方法构建CV-A16 VP1基因的最大分支可信度(MCC)树。还通过贝叶斯天际线图动态分析了CV-A16 VP1基因的进化特征。
所有鉴定出的CV-A16毒株可分为五个主要基因群,分别用GI-GV表示。GIV和GV自2007年以来在中国共同流行,在中国吉林省分离出的CV-A16流行毒株可归类为GIV-3。中国近期流行的CV-A16基因群自2007年以来有共同的祖先。CV-A16 VP1基因的遗传多样性自20世纪90年代中期以来持续增加,1997年和2007年遗传多样性急剧增加,并在2007年达到峰值。2010年之后遗传多样性非常低。CV-A16 VP1基因的进化速率为每年每个位点6.656E-3个替换。
我们预测了动态系统发育趋势,这些趋势表明了CV-A16的爆发趋势,并为由CV-A16引起的手足口病的临床预防和治疗提供了理论基础。