Wang Min, Li Juan, Yao Ming-Xiao, Zhang Ya-Wei, Hu Tao, Carr Michael J, Duchêne Sebastián, Zhang Xing-Cheng, Zhang Zhen-Jie, Zhou Hong, Tong Yi-Gang, Ding Shu-Jun, Wang Xian-Jun, Shi Wei-Feng
Key Laboratory of Etiology and Epidemiology of Emerging Infectious Diseases in Universities of Shandong, Taishan Medical College, Tai'an, China.
Shandong Provincial Key Laboratory of Communicable Disease Control and Prevention, Institute for Viral Disease Control and Prevention, Shandong Center for Disease Control and Prevention, Jinan, China.
Front Microbiol. 2019 May 7;10:1001. doi: 10.3389/fmicb.2019.01001. eCollection 2019.
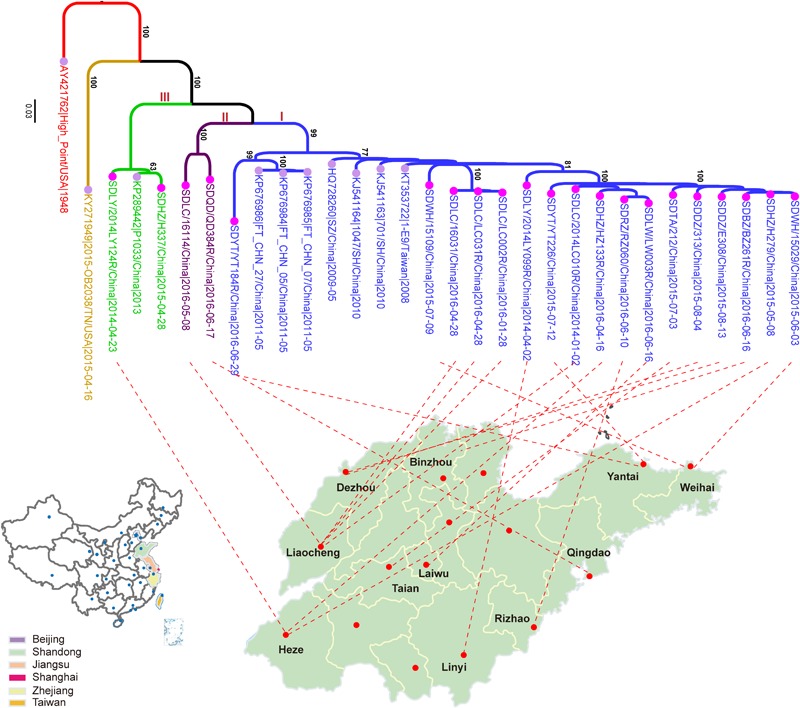
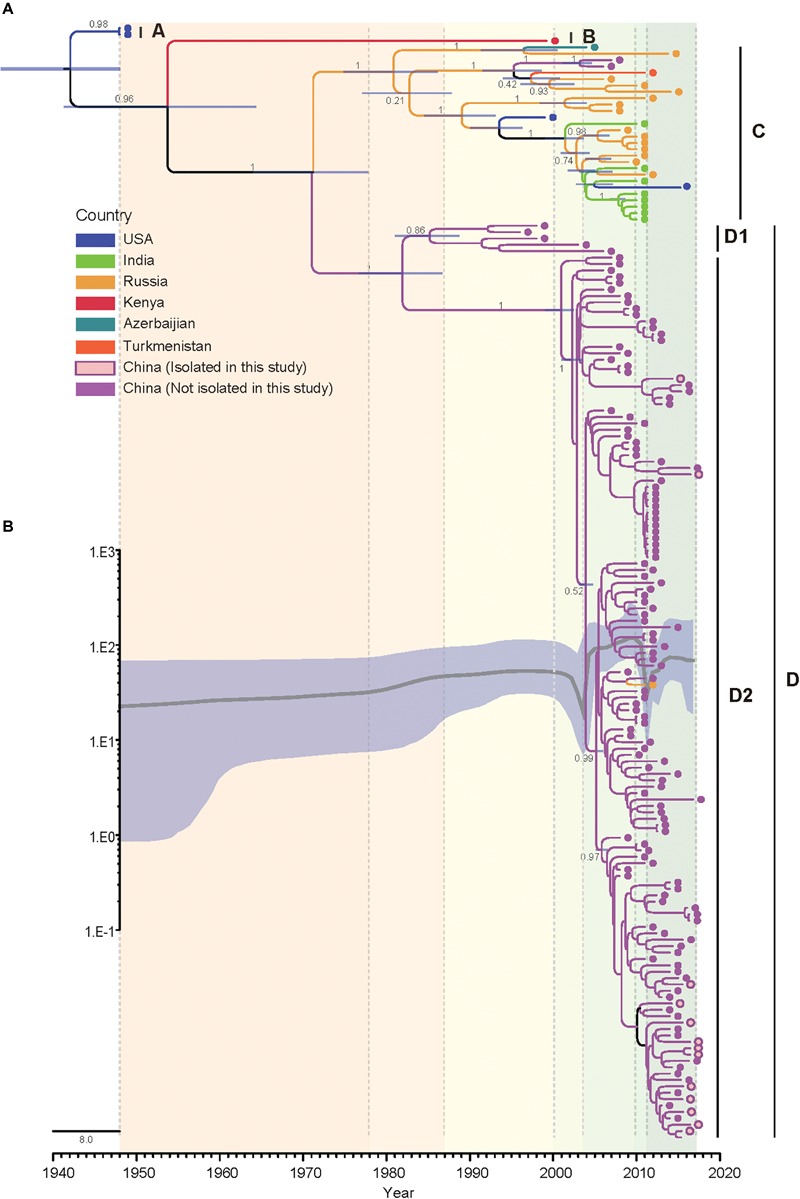
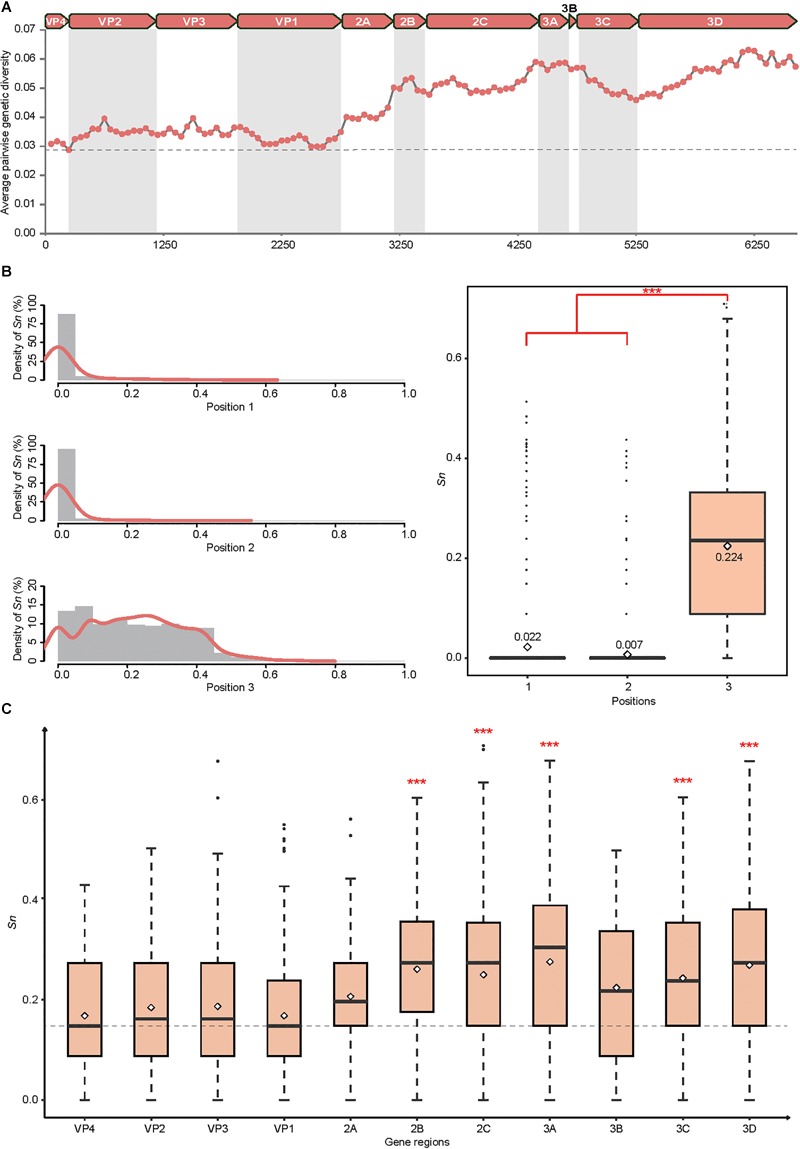
Coxsackievirus A4 (CVA4) is one of the most prevalent pathogens associated with hand, foot and mouth disease (HFMD), an acute febrile illness in children, and is also associated with acute localized exanthema, myocarditis, hepatitis and pancreatitis. Despite this, limited CVA4 genome sequences are currently available. Herein, complete genome sequences from CVA4 strains ( = 21), isolated from patients with HFMD in Shandong province, China between 2014 and 2016, were determined and phylogenetically characterized. Phylogenetic analysis of the gene from a larger CVA4 collection ( = 175) showed that CVA4 has evolved into four separable genotypes: A, B, C, and D; and genotype D could be further classified in to two sub-genotypes: D1 and D2. Each of the 21 newly described genomes derived from isolates that segregated with sub-genotype D2. The CVA4 genomes displayed significant intra-genotypic genetic diversity with frequent synonymous substitutions occurring at the third codon positions, particularly within the P2 region. However, was relatively stable and therefore represents a potential target for molecular diagnostics assays and also for the rational design of vaccine epitopes. The substitution rate of was estimated to be 5.12 × 10 substitutions/site/year, indicative of ongoing CVA4 evolution. Mutations at amino acid residue 169 in gene may be responsible for differing virulence of CVA4 strains. Bayesian skyline plot analysis showed that the population size of CVA4 has experienced several dynamic fluctuations since 1948. In summary, we describe the phylogenetic and molecular characterization of 21 complete genomes from CVA4 isolates which greatly enriches the known genomic diversity of CVA4 and underscores the need for further surveillance of CVA4 in China.
柯萨奇病毒A4(CVA4)是与手足口病(HFMD)相关的最常见病原体之一,手足口病是一种儿童急性发热性疾病,还与急性局限性皮疹、心肌炎、肝炎和胰腺炎有关。尽管如此,目前可用的CVA4基因组序列有限。在此,我们测定并分析了2014年至2016年间从中国山东省手足口病患者中分离出的21株CVA4毒株的全基因组序列,并进行了系统发育特征分析。对来自更大规模CVA4毒株集合(175株)的VP1基因进行系统发育分析表明,CVA4已进化为四个可分离的基因型:A、B、C和D;基因型D可进一步分为两个亚基因型:D1和D2。新描述的21个基因组均来自与亚基因型D2聚类的分离株。CVA4基因组在基因型内显示出显著的遗传多样性,第三密码子位置频繁出现同义替换,特别是在P2区域。然而,VP1相对稳定,因此是分子诊断检测以及疫苗表位合理设计的潜在靶点。估计VP1的替换率为5.12×10-4替换/位点/年,表明CVA4在持续进化。VP1基因中第169位氨基酸残基的突变可能是CVA4毒株毒力不同的原因。贝叶斯天际线图分析表明,自1948年以来,CVA4的种群规模经历了几次动态波动。总之,我们描述了21株CVA4分离株全基因组的系统发育和分子特征,这极大地丰富了已知的CVA4基因组多样性,并强调了中国进一步监测CVA4的必要性。