Huang Xin, Chen Xiao-Guang, Armbruster Peter A
Department of Biology, Georgetown University, 37th and O Streets NW, Washington, DC, 20057, USA.
Key Laboratory of Prevention and Control for Emerging Infectious Diseases of Guangdong Higher Institutes, Department of Pathogen Biology, School of Public Health and Tropical Medicine, Southern Medical University, Guangzhou, China.
BMC Genomics. 2016 Jul 27;17:523. doi: 10.1186/s12864-016-2923-8.
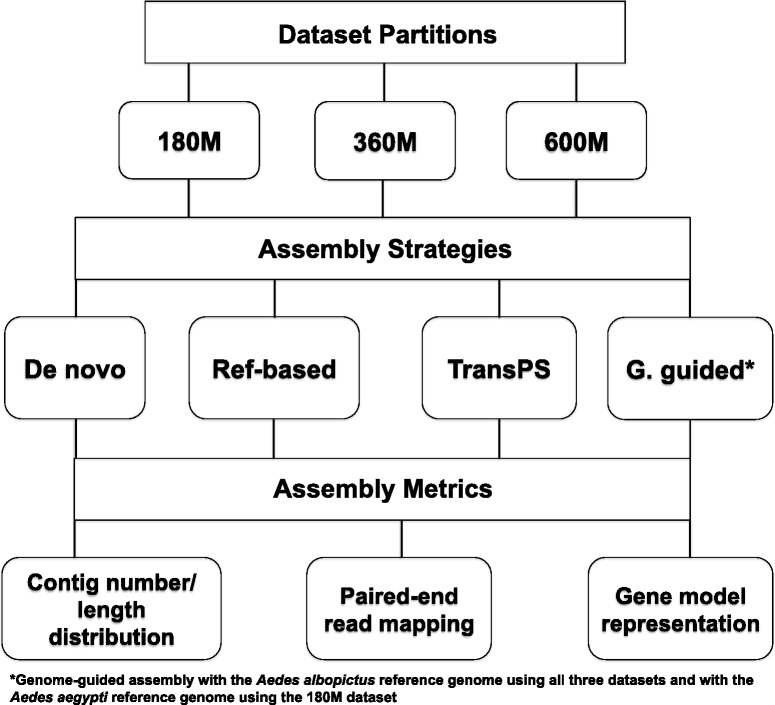
The technological revolution in next-generation sequencing has brought unprecedented opportunities to study any organism of interest at the genomic or transcriptomic level. Transcriptome assembly is a crucial first step for studying the molecular basis of phenotypes of interest using RNA-Sequencing (RNA-Seq). However, the optimal strategy for assembling vast amounts of short RNA-Seq reads remains unresolved, especially for organisms without a sequenced genome. This study compared four transcriptome assembly methods, including a widely used de novo assembler (Trinity), two transcriptome re-assembly strategies utilizing proteomic and genomic resources from closely related species (reference-based re-assembly and TransPS) and a genome-guided assembler (Cufflinks).
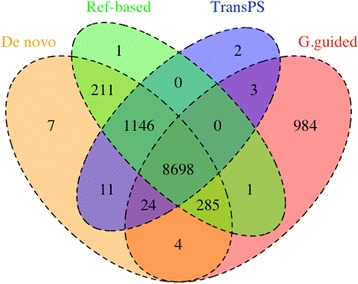
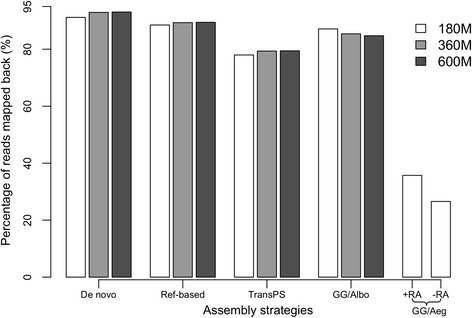
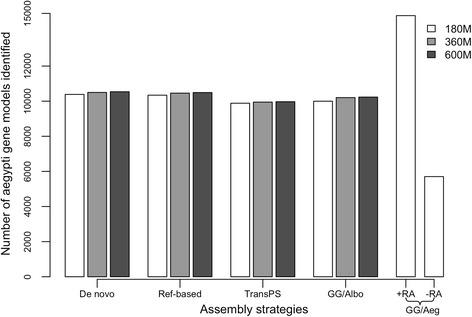
These four assembly strategies were compared using a comprehensive transcriptomic database of Aedes albopictus, for which a genome sequence has recently been completed. The quality of the various assemblies was assessed by the number of contigs generated, contig length distribution, percent paired-end read mapping, and gene model representation via BLASTX. Our results reveal that de novo assembly generates a similar number of gene models relative to genome-guided assembly with a fragmented reference, but produces the highest level of redundancy and requires the most computational power. Using a closely related reference genome to guide transcriptome assembly can generate biased contig sequences. Increasing the number of reads used in the transcriptome assembly tends to increase the redundancy within the assembly and decrease both median contig length and percent identity between contigs and reference protein sequences.
This study provides general guidance for transcriptome assembly of RNA-Seq data from organisms with or without a sequenced genome. The optimal transcriptome assembly strategy will depend upon the subsequent downstream analyses. However, our results emphasize the efficacy of de novo assembly, which can be as effective as genome-guided assembly when the reference genome assembly is fragmented. If a genome assembly and sufficient computational resources are available, it can be beneficial to combine de novo and genome-guided assemblies. Caution should be taken when using a closely related reference genome to guide transcriptome assembly. The quantity of read pairs used in the transcriptome assembly does not necessarily correlate with the quality of the assembly.
新一代测序技术革命为在基因组或转录组水平研究任何感兴趣的生物体带来了前所未有的机遇。转录组组装是使用RNA测序(RNA-Seq)研究感兴趣表型的分子基础的关键第一步。然而,组装大量短RNA-Seq读段的最佳策略仍未解决,尤其是对于没有测序基因组的生物体。本研究比较了四种转录组组装方法,包括一种广泛使用的从头组装器(Trinity)、两种利用来自密切相关物种的蛋白质组和基因组资源的转录组重新组装策略(基于参考的重新组装和TransPS)以及一种基因组引导的组装器(Cufflinks)。
使用白纹伊蚊的综合转录组数据库对这四种组装策略进行了比较,其基因组序列最近已完成。通过生成的重叠群数量、重叠群长度分布、双末端读段映射百分比以及通过BLASTX的基因模型表示来评估各种组装的质量。我们的结果表明,相对于使用片段化参考的基因组引导组装,从头组装产生的基因模型数量相似,但产生的冗余度最高,并且需要最多的计算能力。使用密切相关的参考基因组来指导转录组组装会产生有偏差的重叠群序列。增加转录组组装中使用的读段数量往往会增加组装内的冗余度,并降低重叠群的中位数长度以及重叠群与参考蛋白质序列之间的同一性百分比。
本研究为有或没有测序基因组的生物体的RNA-Seq数据的转录组组装提供了一般性指导。最佳的转录组组装策略将取决于后续的下游分析。然而,我们的结果强调了从头组装的有效性,当参考基因组组装是片段化时,它可以与基因组引导组装一样有效。如果有基因组组装和足够的计算资源,将从头组装和基因组引导组装相结合可能会有好处。在使用密切相关的参考基因组来指导转录组组装时应谨慎。转录组组装中使用的读段对数量不一定与组装质量相关。