Department of Computer Science, Purdue University, West Lafayette, IN, 47907, USA.
Department of Biological Sciences, Purdue University, West Lafayette IN, 47907, USA.
Sci Rep. 2016 Aug 24;6:31725. doi: 10.1038/srep31725.
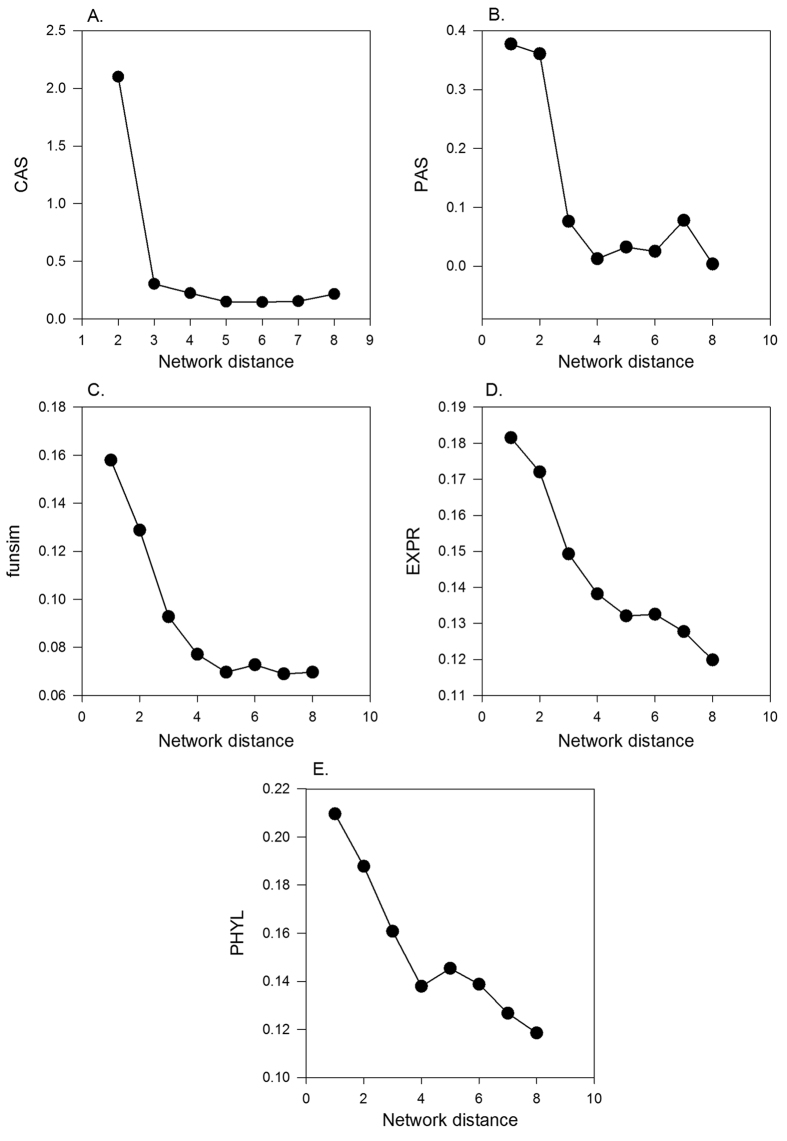
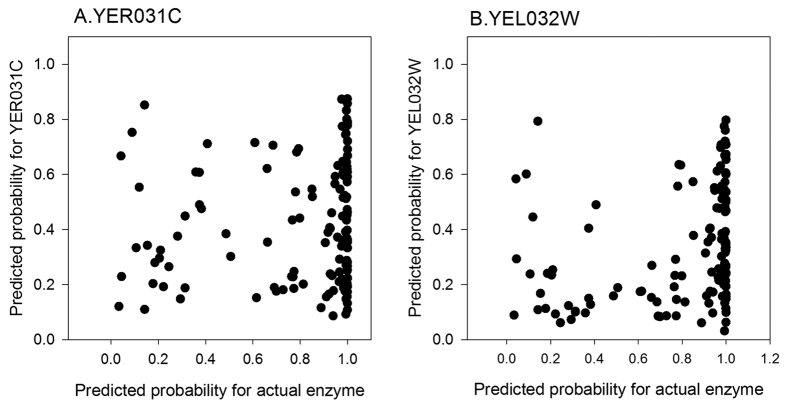
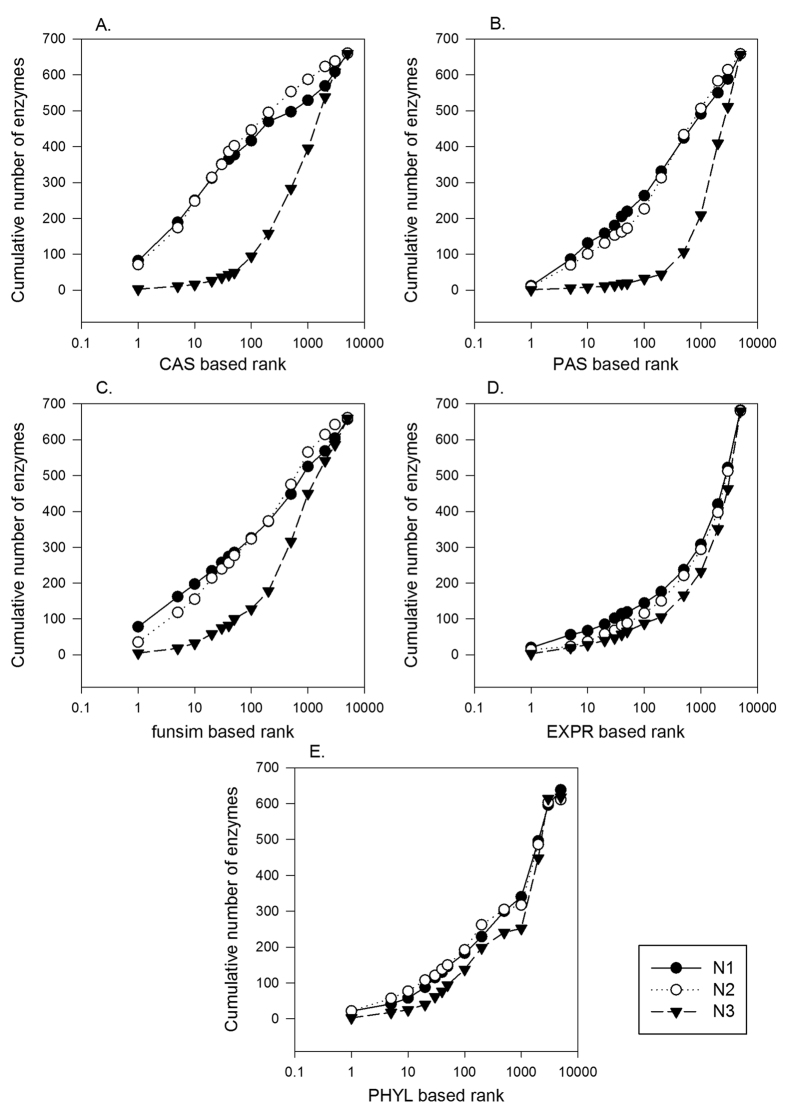
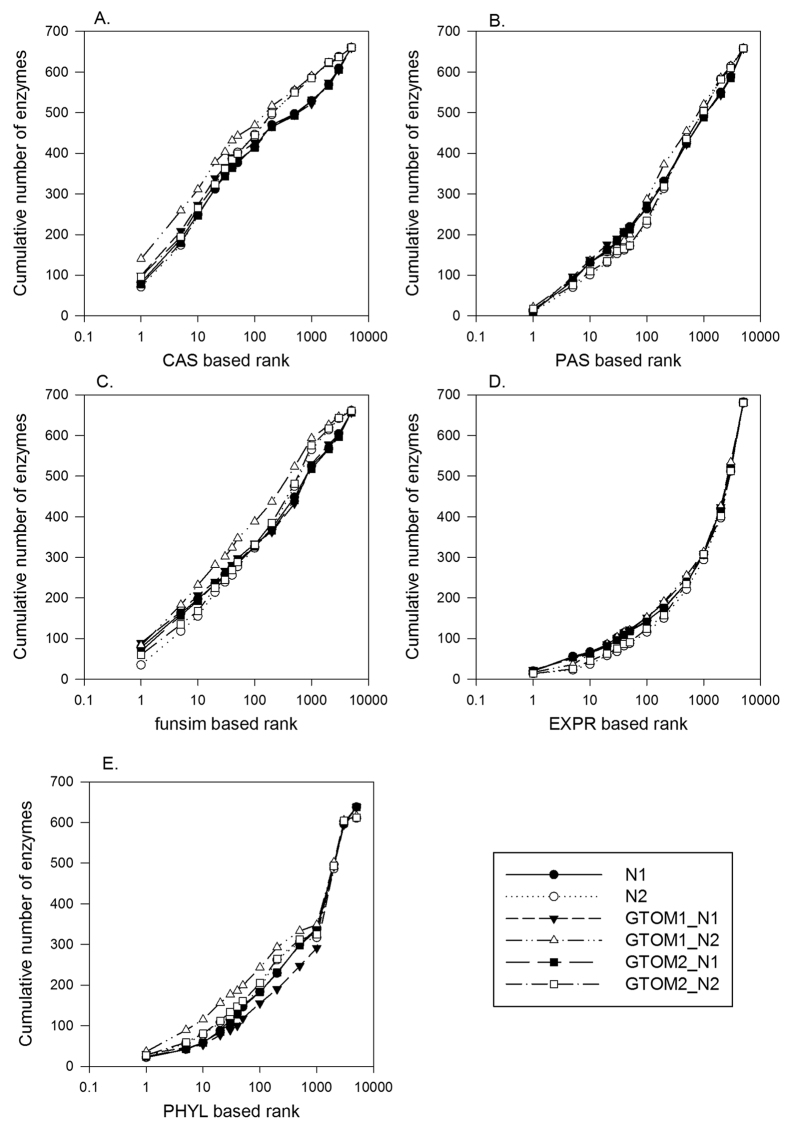
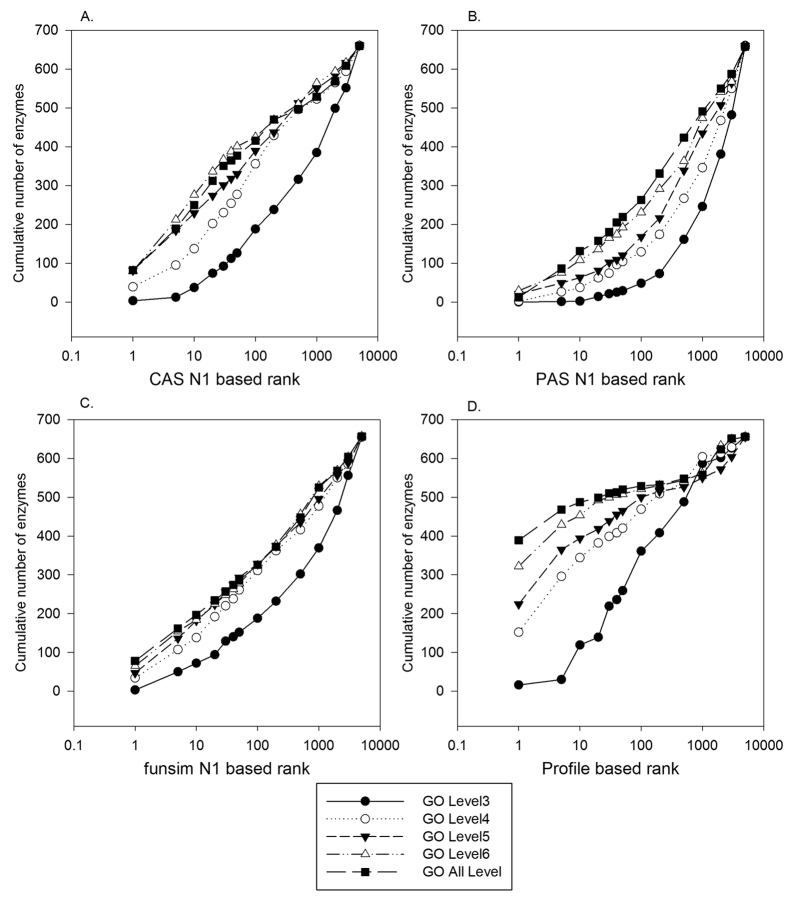
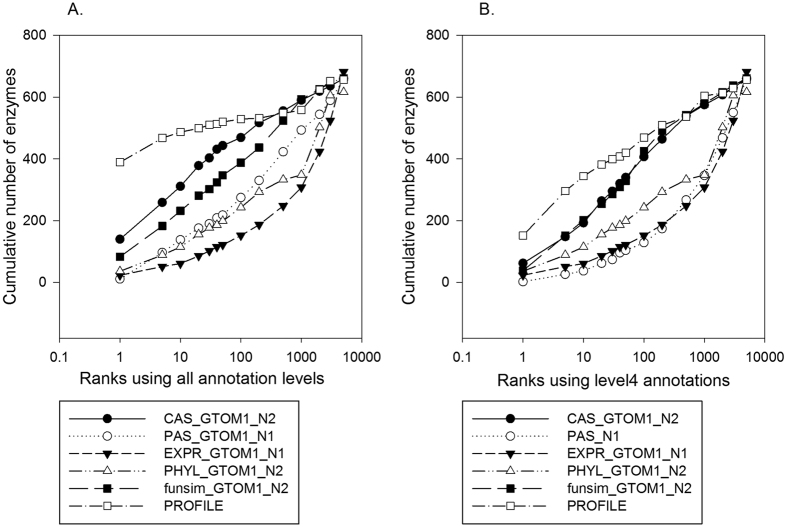
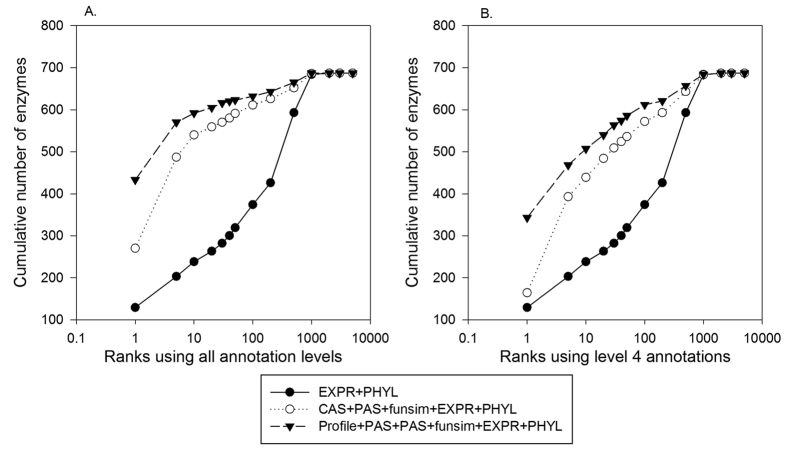
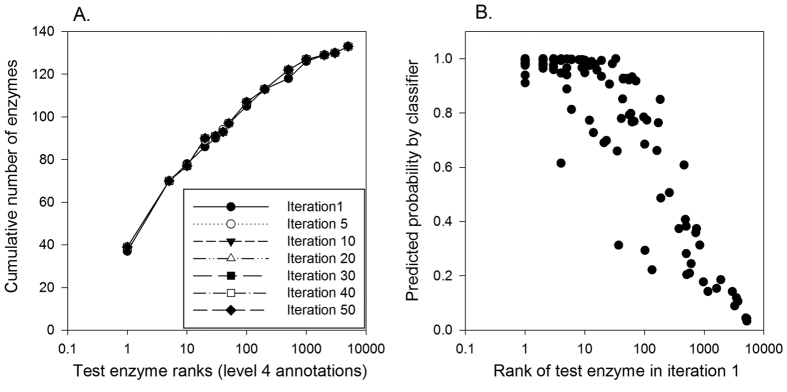
Reconstructing metabolic and signaling pathways is an effective way of interpreting a genome sequence. A challenge in a pathway reconstruction is that often genes in a pathway cannot be easily found, reflecting current imperfect information of the target organism. In this work, we developed a new method for finding missing genes, which integrates multiple features, including gene expression, phylogenetic profile, and function association scores. Particularly, for considering function association between candidate genes and neighboring proteins to the target missing gene in the network, we used Co-occurrence Association Score (CAS) and PubMed Association Score (PAS), which are designed for capturing functional coherence of proteins. We showed that adding CAS and PAS substantially improve the accuracy of identifying missing genes in the yeast enzyme-enzyme network compared to the cases when only the conventional features, gene expression, phylogenetic profile, were used. Finally, it was also demonstrated that the accuracy improves by considering indirect neighbors to the target enzyme position in the network using a proper network-topology-based weighting scheme.
重建代谢和信号通路是解释基因组序列的有效方法。在通路重建中面临的一个挑战是,通路中的基因往往不容易被找到,这反映了目标生物目前不完善的信息。在这项工作中,我们开发了一种新的方法来寻找缺失的基因,该方法整合了多个特征,包括基因表达、系统发育谱和功能关联分数。特别是,为了考虑候选基因与目标缺失基因在网络中的邻近蛋白质之间的功能关联,我们使用了共现关联分数(Co-occurrence Association Score,CAS)和 PubMed 关联分数(PubMed Association Score,PAS),这两种分数是为了捕捉蛋白质的功能一致性而设计的。结果表明,与仅使用传统特征(基因表达、系统发育谱)相比,在酵母酶-酶网络中添加 CAS 和 PAS 可以显著提高识别缺失基因的准确性。最后,还通过使用适当的基于网络拓扑的加权方案来考虑网络中目标酶位置的间接邻居,证明了准确性的提高。