Agricultural Bioinformatics Key Laboratory of Hubei Province, Huazhong Agricultural University, Wuhan 430070, China.
College of Informatics, Huazhong Agricultural University, Wuhan 430070, China.
Sci Rep. 2016 Sep 1;6:32476. doi: 10.1038/srep32476.
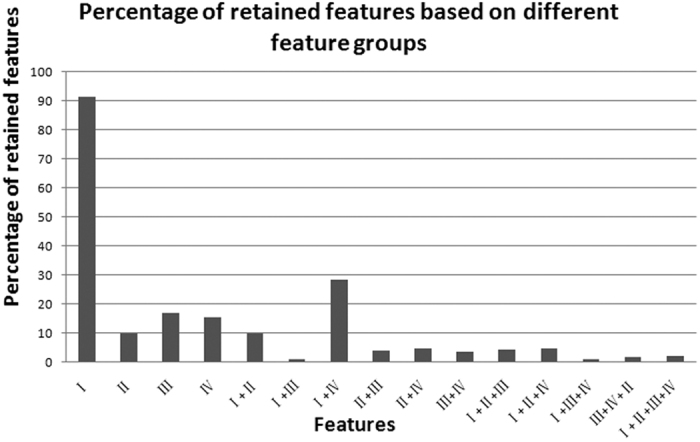
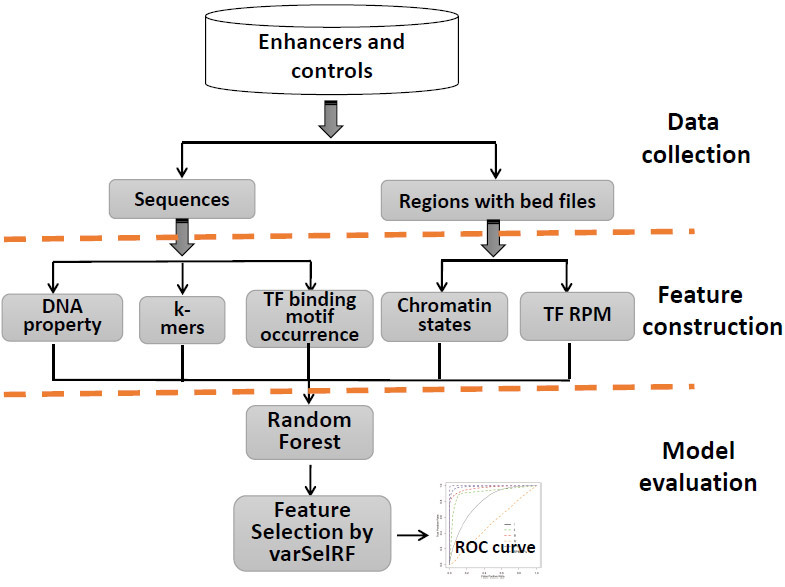
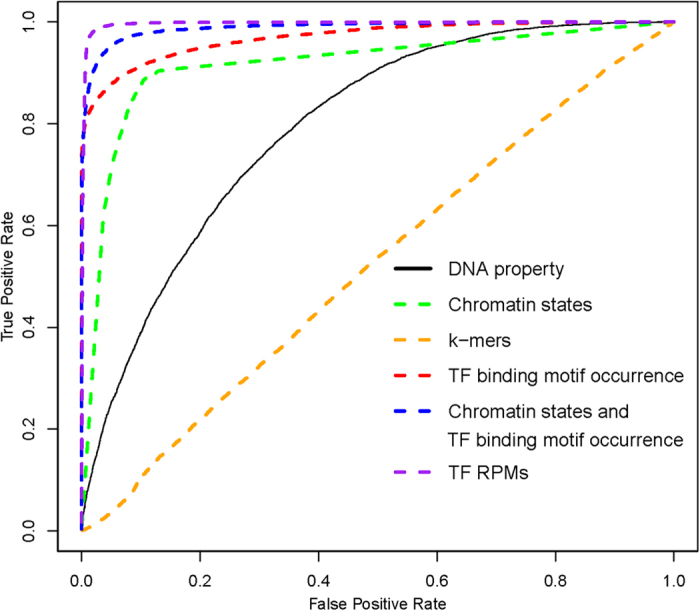
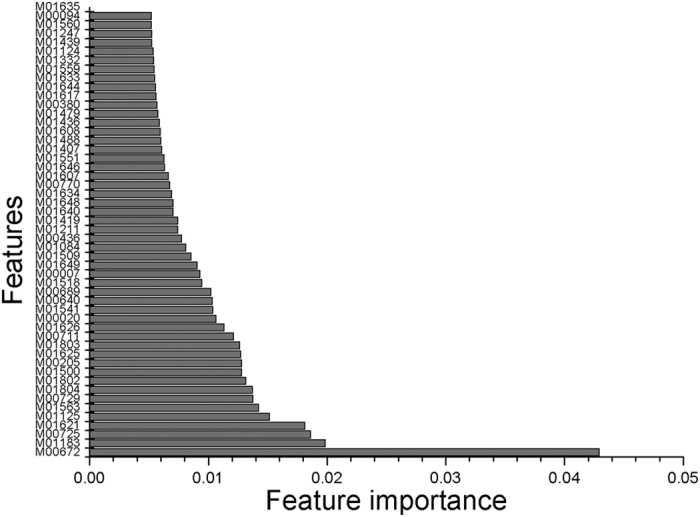
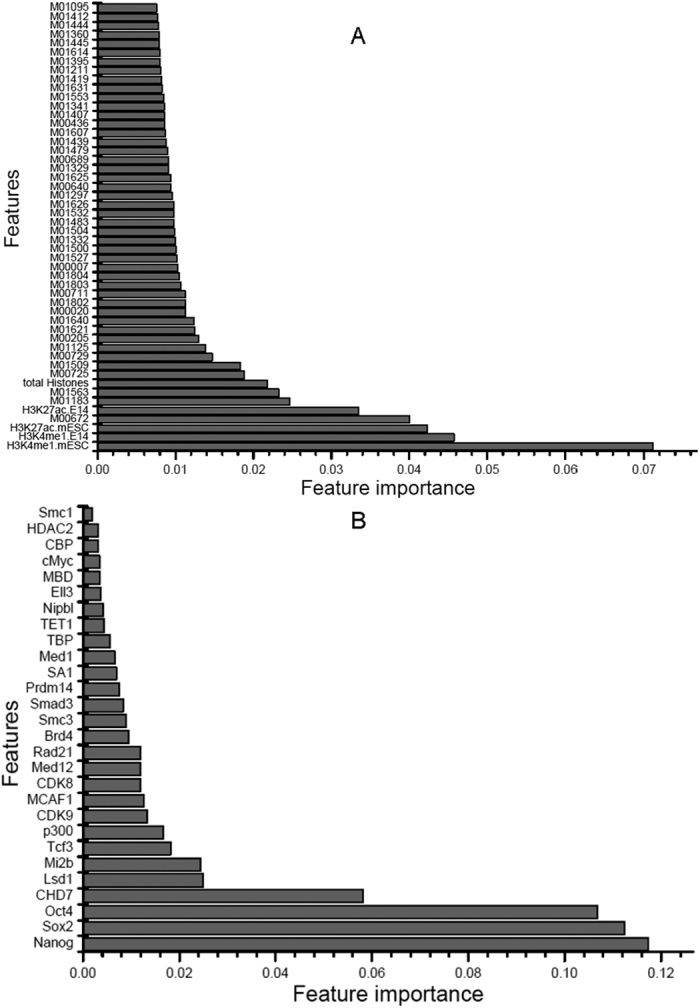
Enhancers interact with gene promoters and form chromatin looping structures that serve important functions in various biological processes, such as the regulation of gene transcription and cell differentiation. However, enhancers are difficult to identify because they generally do not have fixed positions or consensus sequence features, and biological experiments for enhancer identification are costly in terms of labor and expense. In this work, several models were built by using various sequence-based feature sets and their combinations for enhancer prediction. The selected features derived from a recursive feature elimination method showed that the model using a combination of 141 transcription factor binding motif occurrences from 1,422 transcription factor position weight matrices achieved a favorably high prediction accuracy superior to that of other reported methods. The models demonstrated good prediction accuracy for different enhancer datasets obtained from different cell lines/tissues. In addition, prediction accuracy was further improved by integration of chromatin state features. Our method is complementary to wet-lab experimental methods and provides an additional method to identify enhancers.
增强子与基因启动子相互作用,形成染色质环结构,在基因转录调控和细胞分化等各种生物学过程中发挥重要功能。然而,增强子很难识别,因为它们通常没有固定的位置或一致的序列特征,并且增强子的生物学实验在劳动力和费用方面都很昂贵。在这项工作中,我们使用了各种基于序列的特征集及其组合来构建了几个模型,用于增强子预测。从 1,422 个转录因子位置权重矩阵中提取的 141 个转录因子结合基序出现的选择特征表明,使用组合的模型具有优于其他报道方法的有利的高预测准确性。这些模型对来自不同细胞系/组织的不同增强子数据集具有良好的预测准确性。此外,通过整合染色质状态特征,预测准确性进一步提高。我们的方法是对湿实验室实验方法的补充,并提供了一种额外的识别增强子的方法。