Sunseri Jocelyn, Ragoza Matthew, Collins Jasmine, Koes David Ryan
Department of Computational and Systems Biology, School of Medicine, University of Pittsburgh, Suite 3064, Biomedical Science Tower 3 (BST3), 3501 Fifth Avenue, Pittsburgh, PA, 15260, USA.
J Comput Aided Mol Des. 2016 Sep;30(9):761-771. doi: 10.1007/s10822-016-9960-x. Epub 2016 Sep 3.
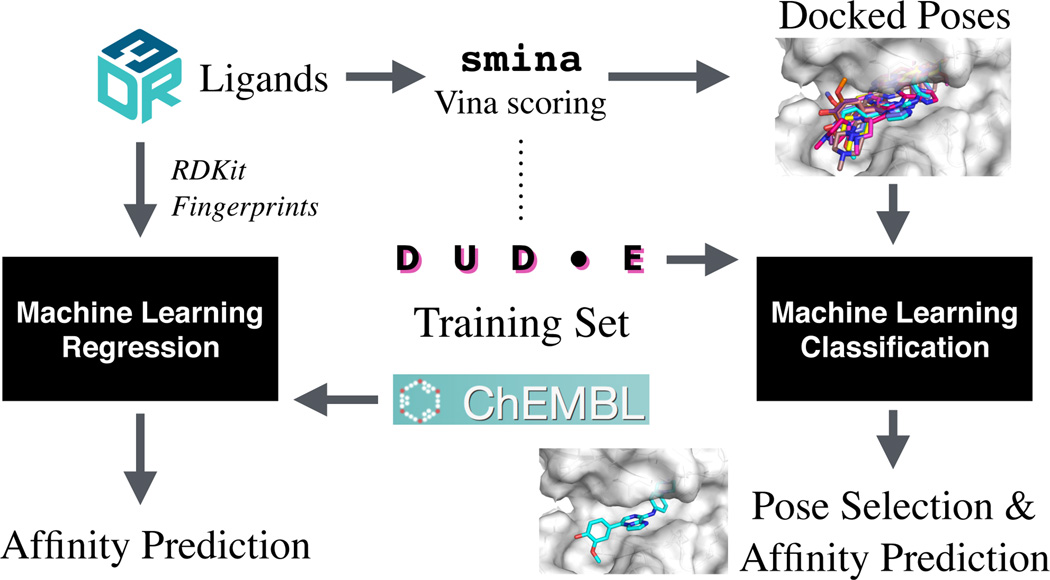
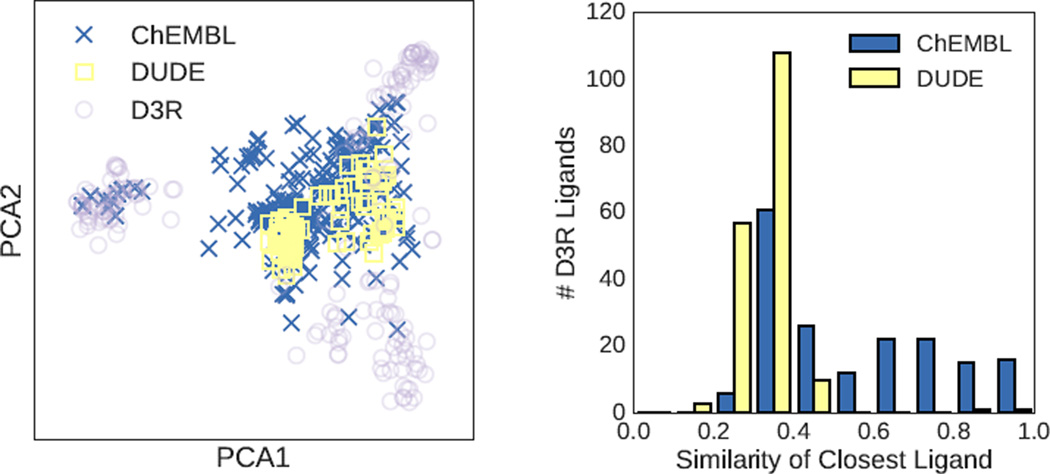
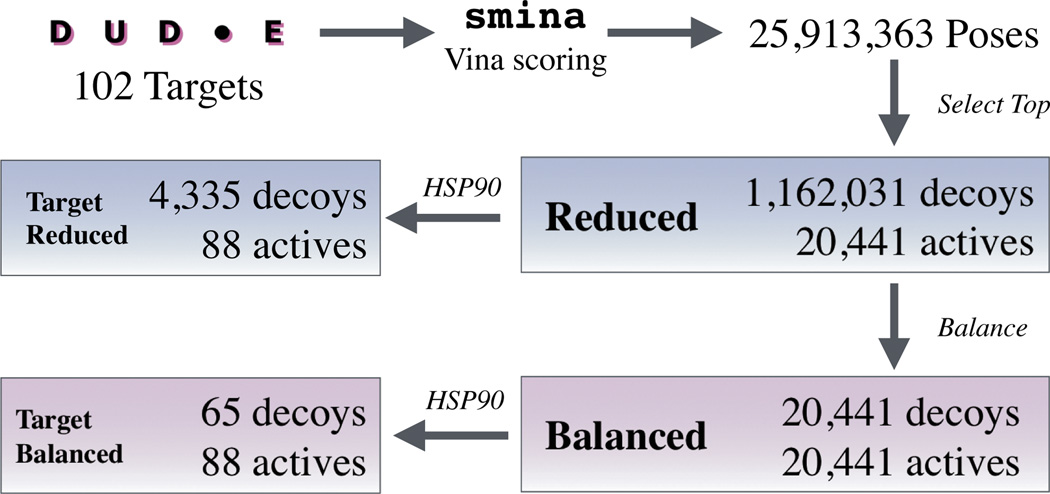
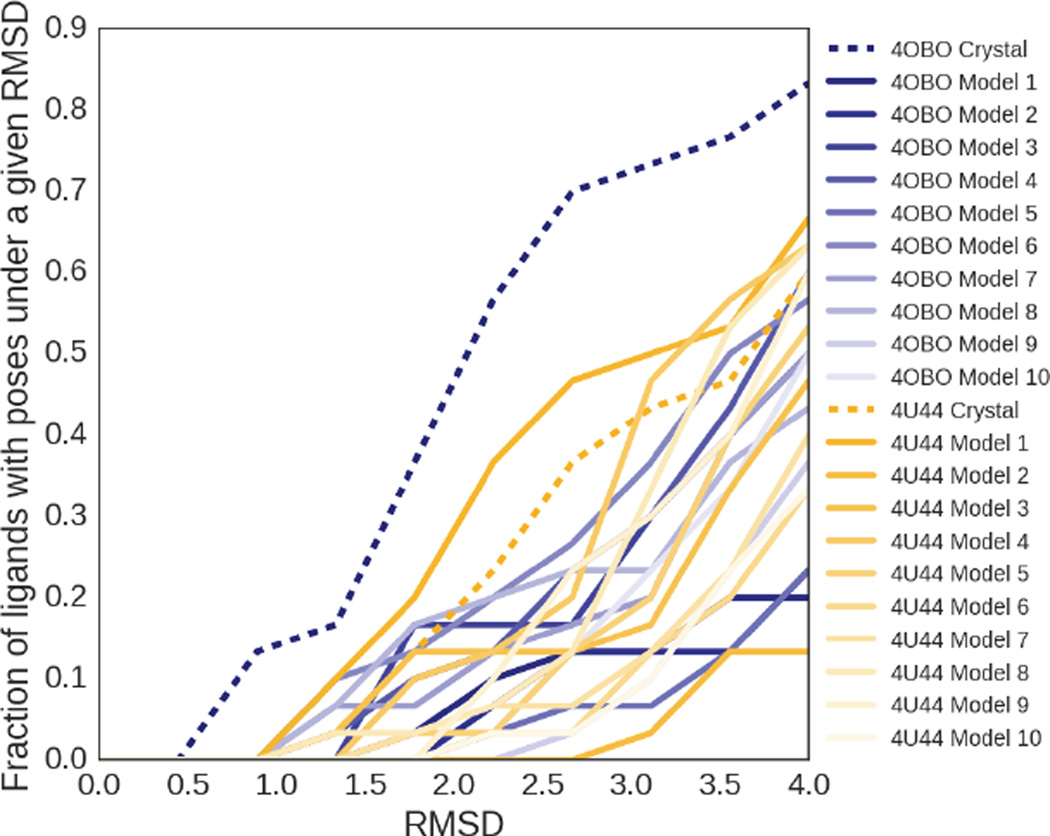
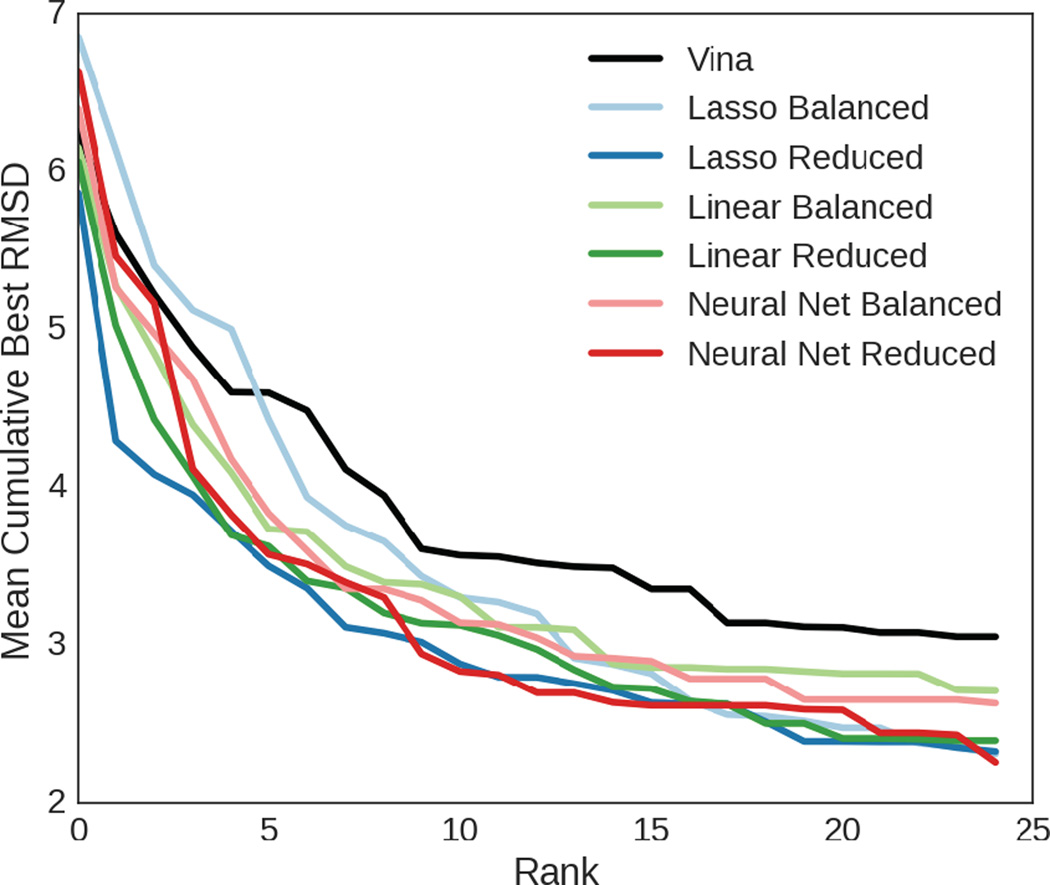
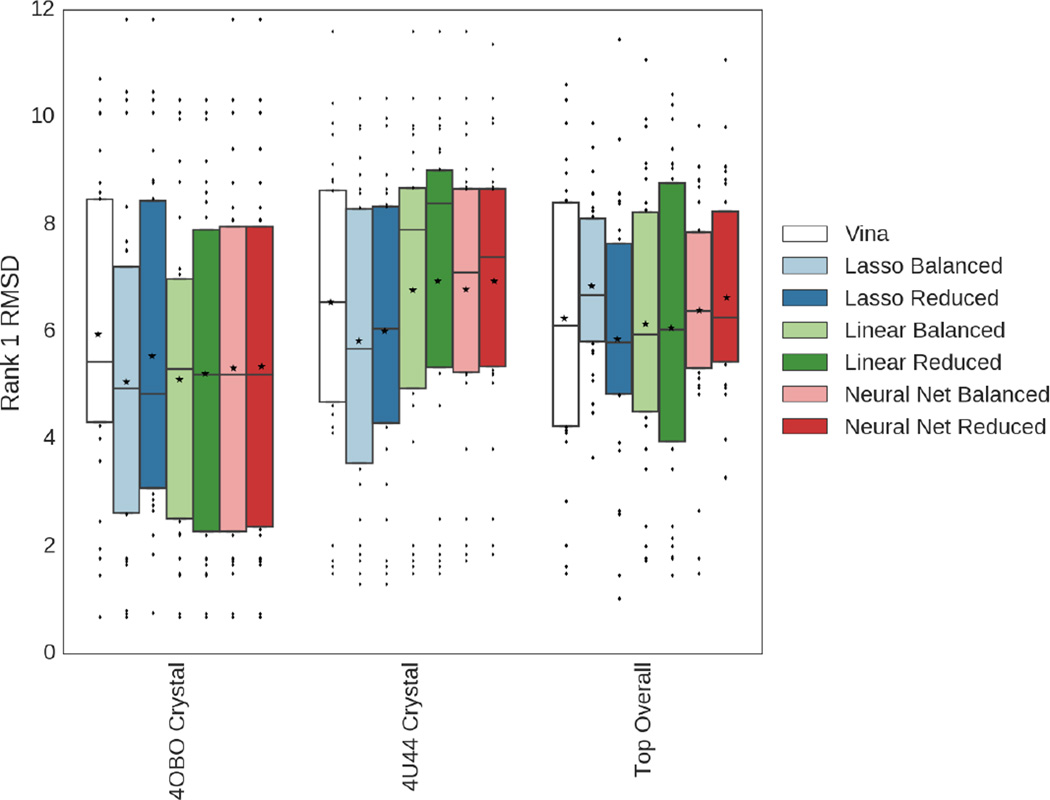
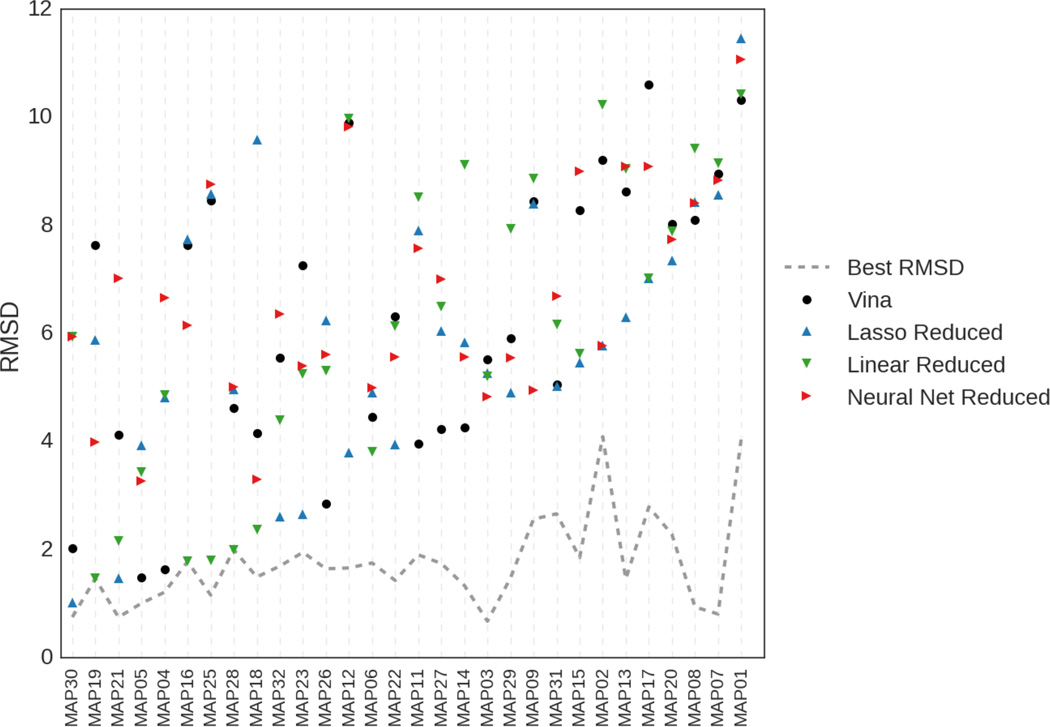
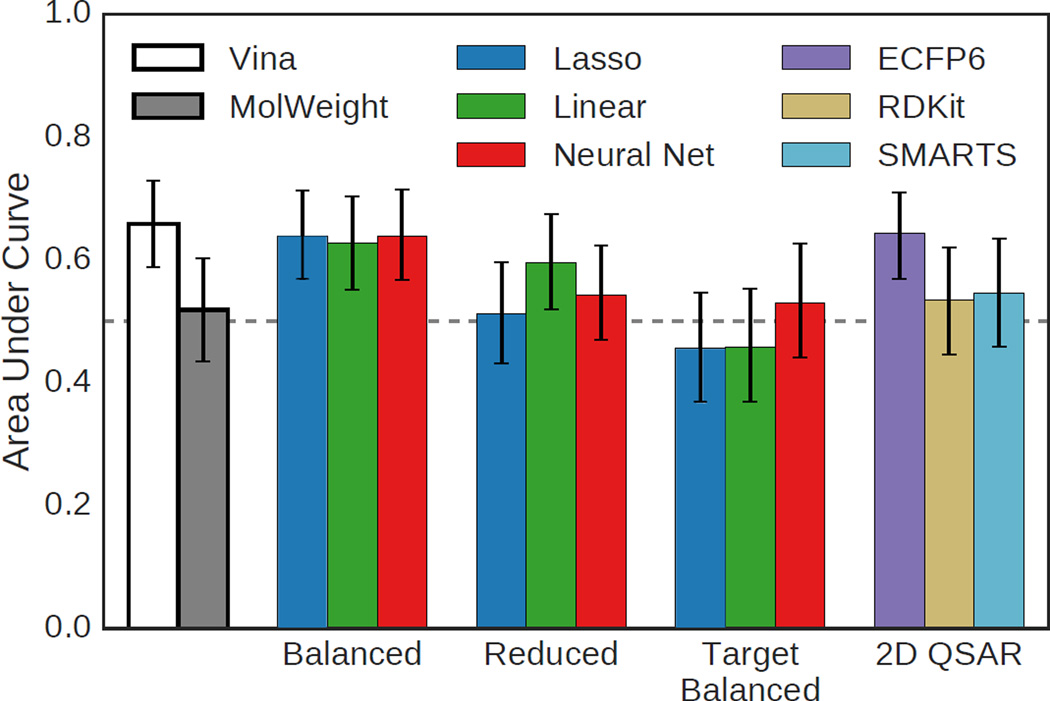
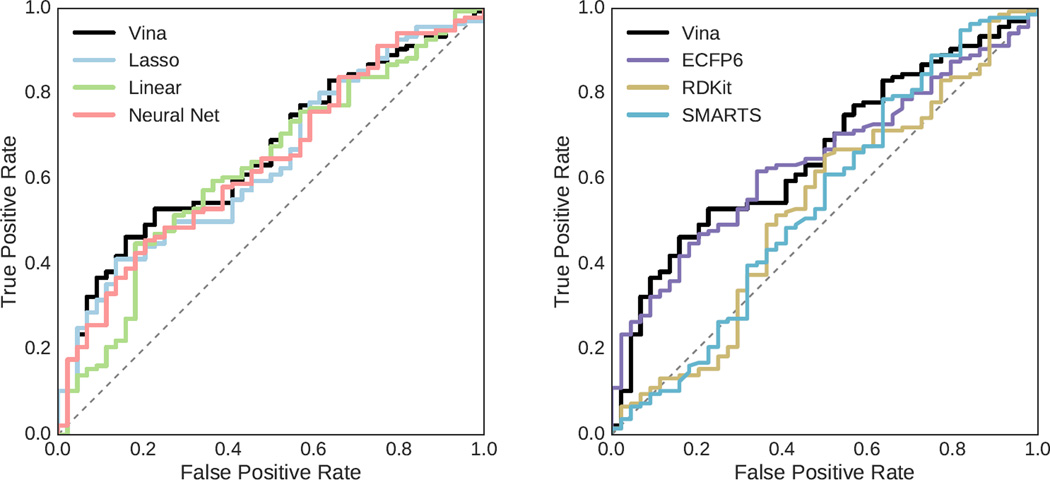
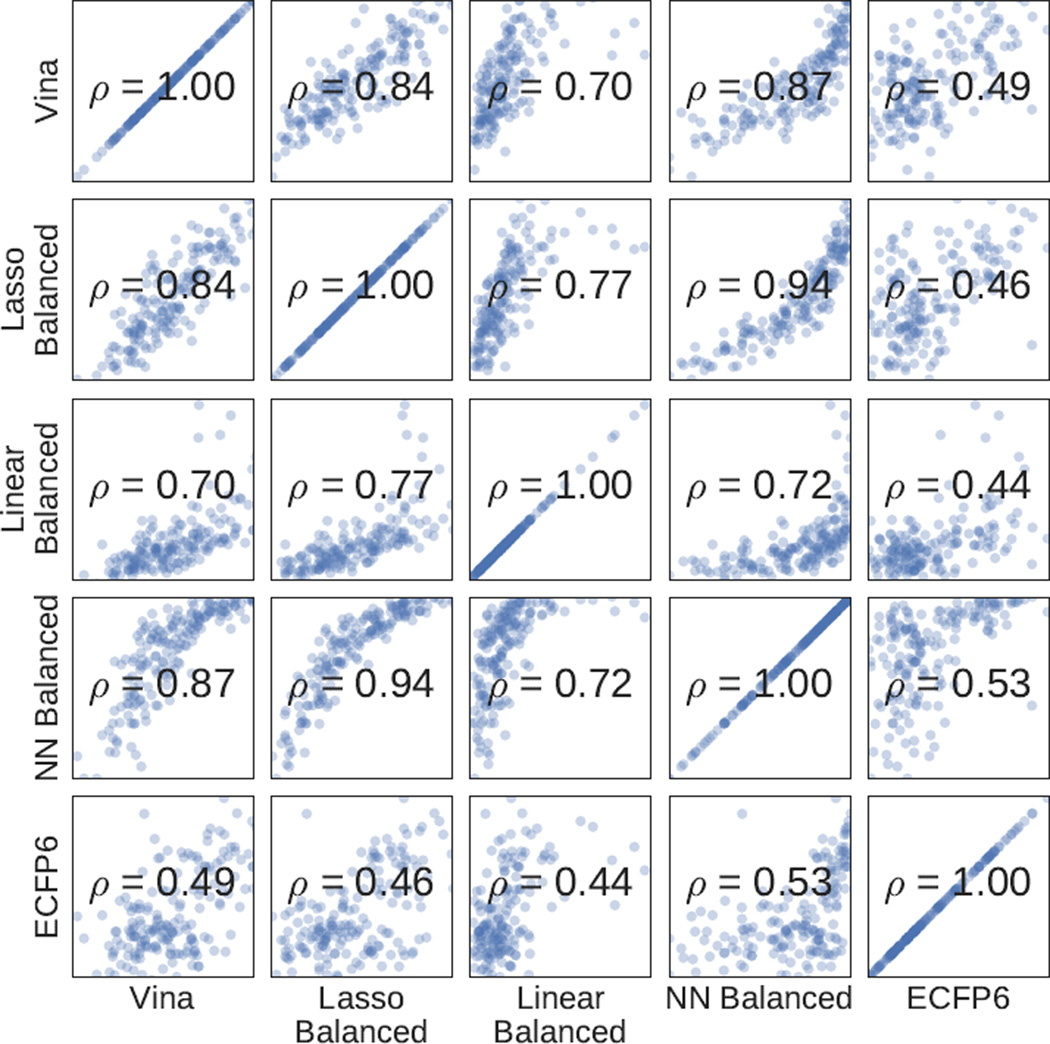
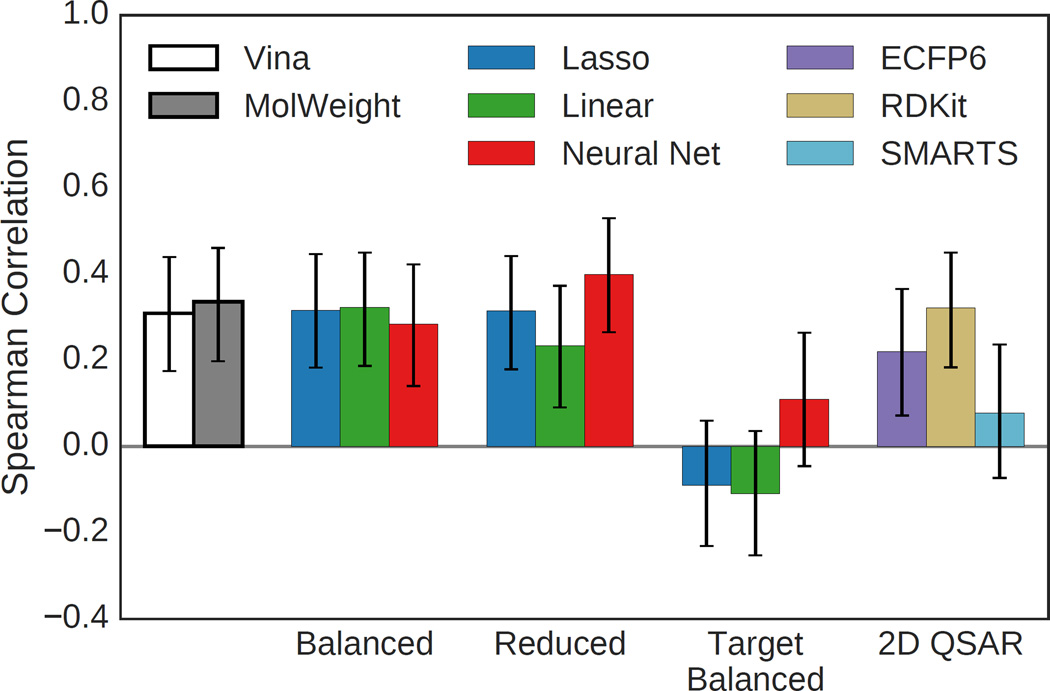
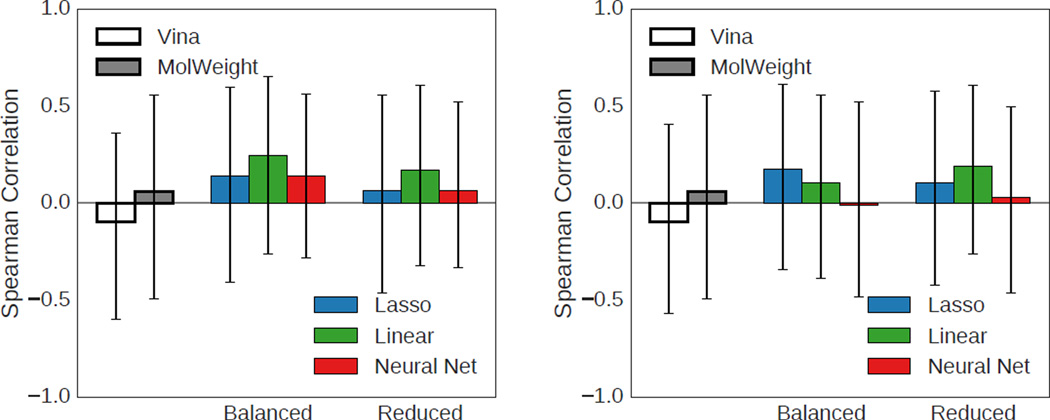
We assess the performance of several machine learning-based scoring methods at protein-ligand pose prediction, virtual screening, and binding affinity prediction. The methods and the manner in which they were trained make them sufficiently diverse to evaluate the utility of various strategies for training set curation and binding pose generation, but they share a novel approach to classification in the context of protein-ligand scoring. Rather than explicitly using structural data such as affinity values or information extracted from crystal binding poses for training, we instead exploit the abundance of data available from high-throughput screening to approach the problem as one of discriminating binders from non-binders. We evaluate the performance of our various scoring methods in the 2015 D3R Grand Challenge and find that although the merits of some features of our approach remain inconclusive, our scoring methods performed comparably to a state-of-the-art scoring function that was fit to binding affinity data.
我们评估了几种基于机器学习的评分方法在蛋白质-配体构象预测、虚拟筛选和结合亲和力预测方面的性能。这些方法及其训练方式足够多样,能够评估各种训练集筛选和结合构象生成策略的效用,但它们在蛋白质-配体评分的背景下采用了一种新颖的分类方法。我们不是明确使用诸如亲和力值或从晶体结合构象中提取的信息等结构数据进行训练,而是利用高通量筛选中可用的大量数据,将该问题作为区分结合剂和非结合剂的问题来处理。我们在2015年D3R大挑战中评估了各种评分方法的性能,发现尽管我们方法的某些特征的优点尚无定论,但我们的评分方法与基于结合亲和力数据拟合的最先进评分函数表现相当。