Laurie Steve, Fernandez-Callejo Marcos, Marco-Sola Santiago, Trotta Jean-Remi, Camps Jordi, Chacón Alejandro, Espinosa Antonio, Gut Marta, Gut Ivo, Heath Simon, Beltran Sergi
CNAG-CRG, Centre for Genomic Regulation (CRG), Barcelona Institute of Science and Technology (BIST), Barcelona, Spain.
Universitat Pompeu Fabra (UPF), Barcelona, Spain.
Hum Mutat. 2016 Dec;37(12):1263-1271. doi: 10.1002/humu.23114. Epub 2016 Sep 26.
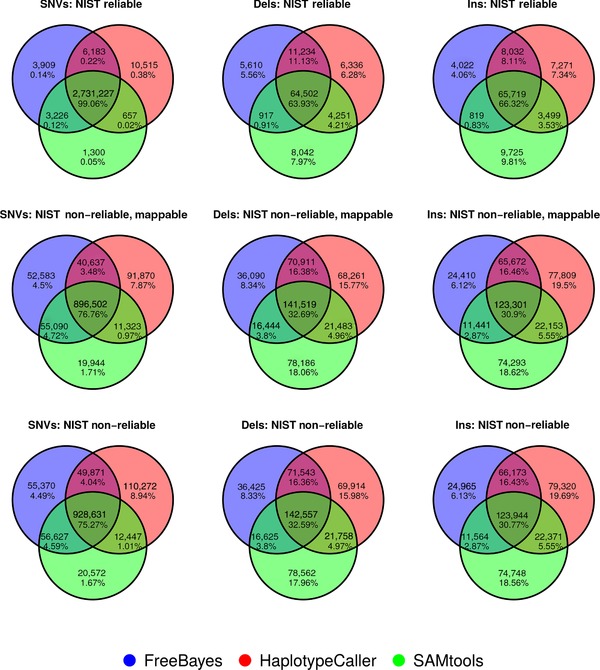
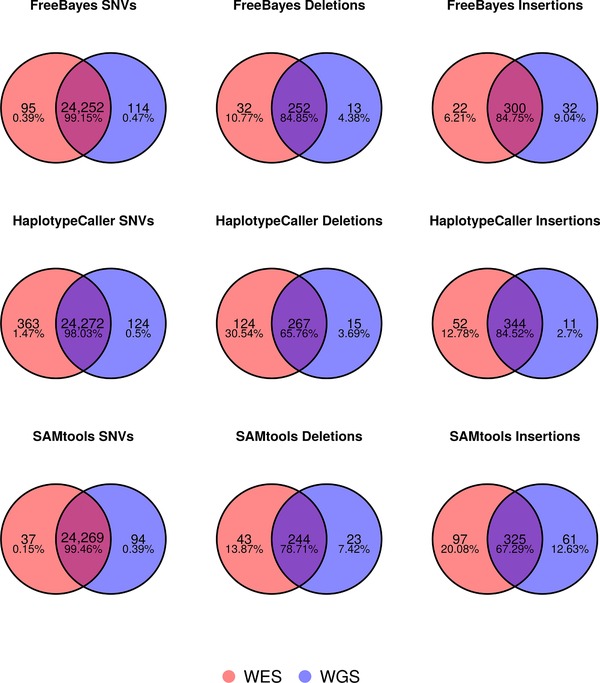
As whole genome sequencing becomes cheaper and faster, it will progressively substitute targeted next-generation sequencing as standard practice in research and diagnostics. However, computing cost-performance ratio is not advancing at an equivalent rate. Therefore, it is essential to evaluate the robustness of the variant detection process taking into account the computing resources required. We have benchmarked six combinations of state-of-the-art read aligners (BWA-MEM and GEM3) and variant callers (FreeBayes, GATK HaplotypeCaller, SAMtools) on whole genome and whole exome sequencing data from the NA12878 human sample. Results have been compared between them and against the NIST Genome in a Bottle (GIAB) variants reference dataset. We report differences in speed of up to 20 times in some steps of the process and have observed that SNV, and to a lesser extent InDel, detection is highly consistent in 70% of the genome. SNV, and especially InDel, detection is less reliable in 20% of the genome, and almost unfeasible in the remaining 10%. These findings will aid in choosing the appropriate tools bearing in mind objectives, workload, and computing infrastructure available.
随着全基因组测序变得更加便宜和快速,它将逐渐取代靶向新一代测序,成为研究和诊断中的标准做法。然而,计算成本效益比并没有以相同的速度提升。因此,考虑到所需的计算资源,评估变异检测过程的稳健性至关重要。我们在来自NA12878人类样本的全基因组和全外显子组测序数据上,对六种最先进的读段比对器(BWA-MEM和GEM3)和变异调用器(FreeBayes、GATK HaplotypeCaller、SAMtools)的组合进行了基准测试。将它们之间的结果以及与美国国家标准与技术研究院(NIST)基因组瓶中基因组(GIAB)变异参考数据集的结果进行了比较。我们报告了该过程某些步骤中高达20倍的速度差异,并观察到在70%的基因组中,单核苷酸变异(SNV)以及程度较轻的插入缺失(InDel)检测高度一致。在20%的基因组中,SNV尤其是InDel检测不太可靠,而在其余10%的基因组中几乎不可行。这些发现将有助于根据目标、工作量和可用的计算基础设施选择合适的工具。