Kelly Benjamin J, Fitch James R, Hu Yangqiu, Corsmeier Donald J, Zhong Huachun, Wetzel Amy N, Nordquist Russell D, Newsom David L, White Peter
Center for Microbial Pathogenesis, The Research Institute at Nationwide Children's Hospital, 700 Children's Drive, Columbus 43205, OH, USA.
Department of Pediatrics, College of Medicine, The Ohio State University, Columbus, Ohio, USA.
Genome Biol. 2015 Jan 20;16(1):6. doi: 10.1186/s13059-014-0577-x.
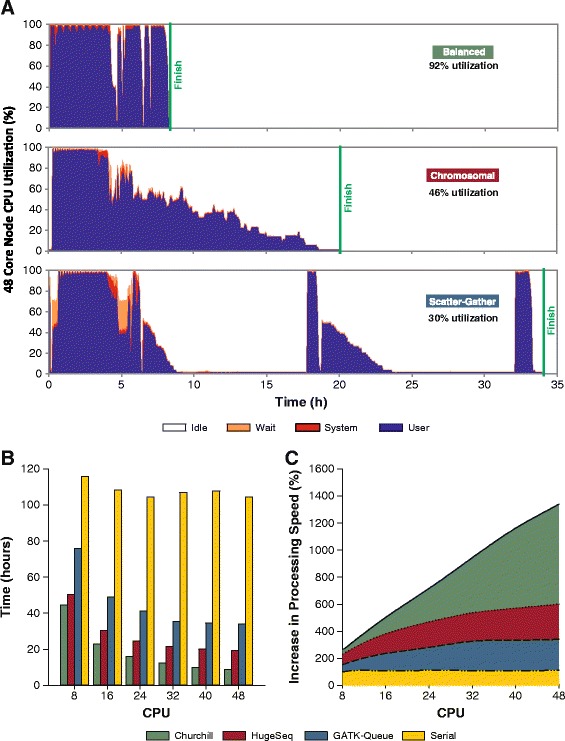
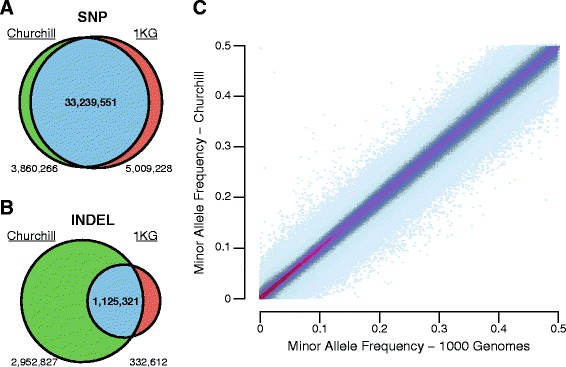
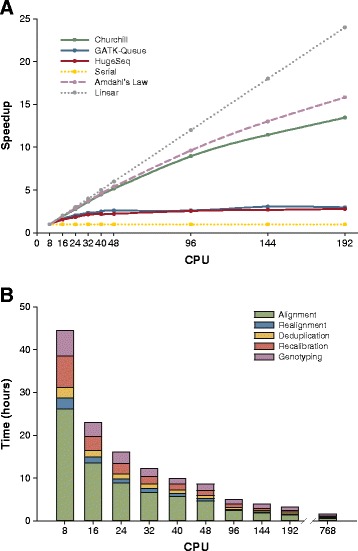
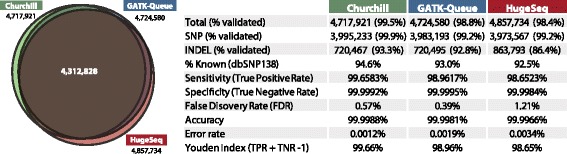
While advances in genome sequencing technology make population-scale genomics a possibility, current approaches for analysis of these data rely upon parallelization strategies that have limited scalability, complex implementation and lack reproducibility. Churchill, a balanced regional parallelization strategy, overcomes these challenges, fully automating the multiple steps required to go from raw sequencing reads to variant discovery. Through implementation of novel deterministic parallelization techniques, Churchill allows computationally efficient analysis of a high-depth whole genome sample in less than two hours. The method is highly scalable, enabling full analysis of the 1000 Genomes raw sequence dataset in a week using cloud resources. http://churchill.nchri.org/.
虽然基因组测序技术的进步使大规模群体基因组学成为可能,但目前分析这些数据的方法依赖于并行化策略,这些策略扩展性有限、实施复杂且缺乏可重复性。Churchill是一种平衡的区域并行化策略,它克服了这些挑战,将从原始测序读数到变异发现所需的多个步骤完全自动化。通过实施新颖的确定性并行化技术,Churchill能够在不到两小时内对高深度全基因组样本进行高效计算分析。该方法具有高度可扩展性,使用云资源一周内就能对千人基因组原始序列数据集进行全面分析。http://churchill.nchri.org/