Zhou Xiaofan, Peris David, Kominek Jacek, Kurtzman Cletus P, Hittinger Chris Todd, Rokas Antonis
Department of Biological Sciences, Vanderbilt University, Nashville, Tennessee 37235.
Laboratory of Genetics, Genome Center of Wisconsin, Department of Energy Great Lakes Bioenergy Research Center, Wisconsin Energy Institute, J. F. Crow Institute for the Study of Evolution, University of Wisconsin-Madison, Wisconsin 53706.
G3 (Bethesda). 2016 Nov 8;6(11):3655-3662. doi: 10.1534/g3.116.034249.
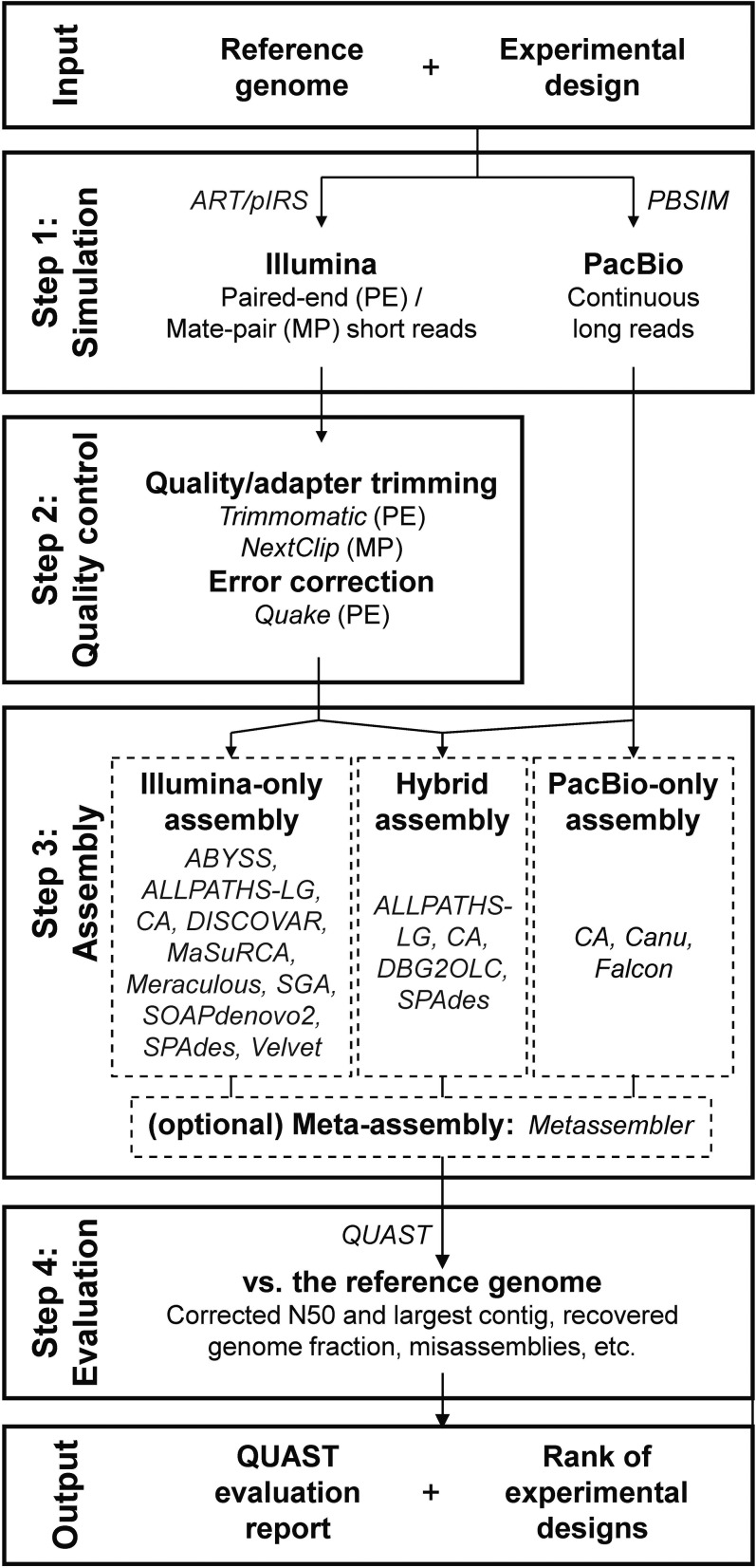
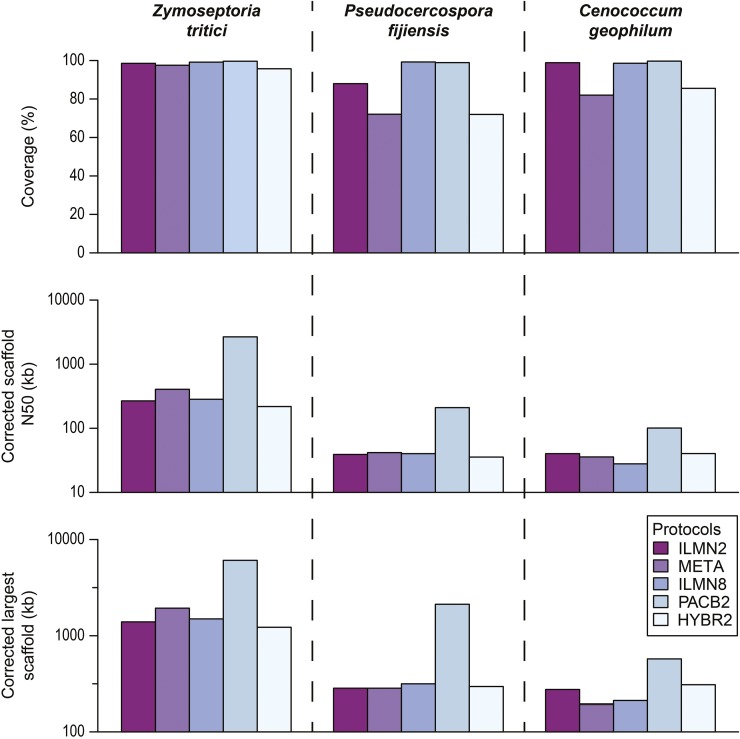
The availability of genomes across the tree of life is highly biased toward vertebrates, pathogens, human disease models, and organisms with relatively small and simple genomes. Recent progress in genomics has enabled the decoding of the genome of virtually any organism, greatly expanding its potential for understanding the biology and evolution of the full spectrum of biodiversity. The increasing diversity of sequencing technologies, assays, and assembly algorithms have augmented the complexity of genome sequencing projects in nonmodel organisms. To reduce the costs and challenges in genome sequencing projects and streamline their experimental design and analysis, we developed iWGS ( hole enome equencer and Analyzer), an automated pipeline for guiding the choice of appropriate sequencing strategy and assembly protocols. iWGS seamlessly integrates the four key steps of a genome sequencing project: data generation (through simulation), data quality control, assembly, and assembly evaluation and validation. The last three steps can also be applied to the analysis of real data. iWGS is designed to enable the user to have great flexibility in testing the range of experimental designs available for genome sequencing projects, and supports all major sequencing technologies and popular assembly tools. Three case studies illustrate how iWGS can guide the design of genome sequencing projects, and evaluate the performance of a wide variety of user-specified sequencing strategies and assembly protocols on genomes of differing architectures. iWGS, along with a detailed documentation, is freely available at https://github.com/zhouxiaofan1983/iWGS.
整个生命之树中基因组的可得性严重偏向于脊椎动物、病原体、人类疾病模型以及基因组相对小而简单的生物。基因组学的最新进展使得几乎任何生物的基因组都能被解码,极大地扩展了其理解全谱生物多样性的生物学和进化的潜力。测序技术、检测方法和组装算法的日益多样化增加了非模式生物基因组测序项目的复杂性。为了降低基因组测序项目的成本和挑战,并简化其实验设计和分析,我们开发了iWGS(全基因组测序仪和分析仪),这是一个用于指导选择合适测序策略和组装协议的自动化流程。iWGS无缝集成了基因组测序项目的四个关键步骤:数据生成(通过模拟)、数据质量控制、组装以及组装评估和验证。最后三个步骤也可应用于真实数据的分析。iWGS旨在让用户在测试基因组测序项目可用的实验设计范围时具有很大的灵活性,并支持所有主要的测序技术和流行的组装工具。三个案例研究说明了iWGS如何指导基因组测序项目的设计,并评估各种用户指定的测序策略和组装协议在不同结构基因组上的性能。iWGS以及详细文档可在https://github.com/zhouxiaofan1983/iWGS上免费获取。