Carpenter Colleen, Sorenson Roderick J, Jin Yafei, Klossowski Szymon, Cierpicki Tomasz, Gnegy Margaret, Showalter Hollis D
Department of Pharmacology, University of Michigan, Ann Arbor, MI 48109, United States.
Department of Medicinal Chemistry, University of Michigan, Ann Arbor, MI 48109, United States; Vahlteich Medicinal Chemistry Core, University of Michigan, Ann Arbor, MI 48109, United States.
Bioorg Med Chem. 2016 Nov 1;24(21):5495-5504. doi: 10.1016/j.bmc.2016.09.002. Epub 2016 Sep 4.
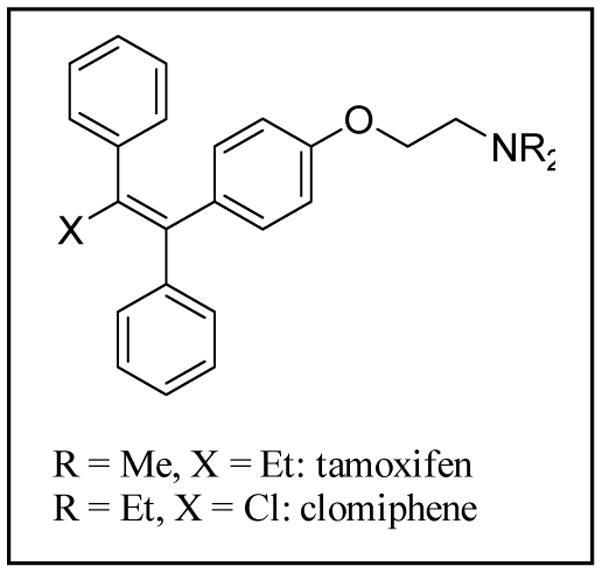
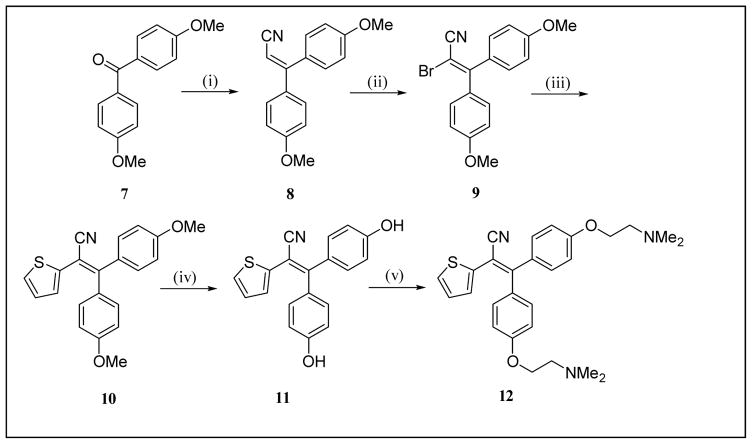
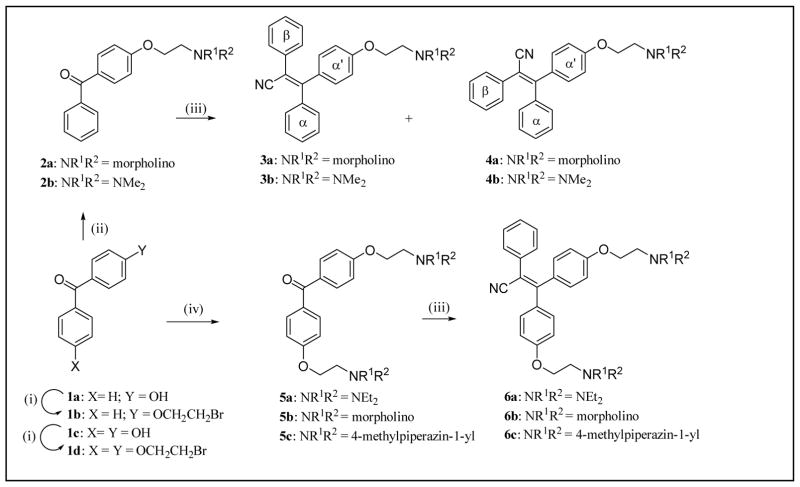
The clinical selective estrogen receptor modulator tamoxifen is also a modest inhibitor of protein kinase C, a target implicated in several untreatable brain diseases such as amphetamine abuse. This inhibition and tamoxifen's ability to cross the blood brain barrier make it an attractive scaffold to conduct further SAR studies toward uncovering effective therapies for such diseases. Utilizing the known compound 6a as a starting template and guided by computational tools to derive physicochemical properties known to be important for CNS permeable drugs, the design and synthesis of a small series of novel triarylacrylonitrile analogues have been carried out providing compounds with enhanced potency and selectivity for PKC over the estrogen receptor relative to tamoxifen. Shortened synthetic routes compared to classical procedures have been developed for analogues incorporating a β-phenyl ring, which involve installing dialkylaminoalkoxy side chains first off the α and/or α' rings of a precursor benzophenone and then condensing the resultant ketones with phenylacetonitrile anion. A second novel, efficient and versatile route utilizing Suzuki chemistry has also been developed, which will allow for the introduction of a wide range of β-aryl or β-heteroaryl moieties and side-chain substituents onto the acrylonitrile core. For analogues possessing a single side chain off the α- or α'-ring, novel 2D NMR experiments have been carried out that allow for unambiguous assignment of E- and Z-stereochemistry. From the SAR analysis, one compound, 6c, shows markedly increased potency and selectivity for inhibiting PKC with an IC of 80nM for inhibition of PKC protein substrate and >10μM for binding to the estrogen receptor α (tamoxifen IC=20μM and 222nM, respectively). The data on 6c provide support for further exploration of PKC as a druggable target for the treatment of amphetamine abuse.
临床选择性雌激素受体调节剂他莫昔芬也是蛋白激酶C的适度抑制剂,蛋白激酶C是涉及多种难以治疗的脑部疾病(如苯丙胺滥用)的一个靶点。这种抑制作用以及他莫昔芬穿越血脑屏障的能力,使其成为一个有吸引力的骨架,可用于开展进一步的构效关系(SAR)研究,以发现针对此类疾病的有效疗法。以已知化合物6a作为起始模板,并在计算工具的指导下,得出对中枢神经系统渗透性药物很重要的物理化学性质,开展了一小系列新型三芳基丙烯腈类似物的设计与合成,得到了相对于他莫昔芬而言对蛋白激酶C具有更高效力和选择性的化合物,且对雌激素受体的选择性更高。与经典方法相比,已开发出用于含有β-苯环的类似物的缩短合成路线,该路线包括首先在前体二苯甲酮的α和/或α'环上引入二烷基氨基烷氧基侧链,然后将所得酮与苯乙腈阴离子缩合。还开发了第二条利用铃木化学的新颖、高效且通用的路线,该路线将允许在丙烯腈核心上引入多种β-芳基或β-杂芳基部分以及侧链取代基。对于在α-或α'-环上具有单个侧链的类似物,已开展了新型二维核磁共振实验,能够明确确定E-和Z-立体化学。从构效关系分析来看,一种化合物6c对抑制蛋白激酶C显示出显著提高的效力和选择性,抑制蛋白激酶C蛋白底物的IC为80nM,与雌激素受体α结合的IC>10μM(他莫昔芬的IC分别为20μM和222nM)。关于6c的数据为进一步探索将蛋白激酶C作为治疗苯丙胺滥用的可成药靶点提供了支持。