de Kovel Carolien G F, Brilstra Eva H, van Kempen Marjan J A, Van't Slot Ruben, Nijman Isaac J, Afawi Zaid, De Jonghe Peter, Djémié Tania, Guerrini Renzo, Hardies Katia, Helbig Ingo, Hendrickx Rik, Kanaan Moine, Kramer Uri, Lehesjoki Anna-Elina E, Lemke Johannes R, Marini Carla, Mei Davide, Møller Rikke S, Pendziwiat Manuela, Stamberger Hannah, Suls Arvid, Weckhuysen Sarah, Koeleman Bobby P C
Department of Genetics UMC Utrecht Utrecht The Netherlands.
Tel Aviv Sourasky Medical Center6 Weizmann St.Tel AvivIsrael; Genetics of Epilepsy Research in Israel Tel-Aviv University Medical SchoolTel-AvivIsrael.
Mol Genet Genomic Med. 2016 Jul 30;4(5):568-80. doi: 10.1002/mgg3.235. eCollection 2016 Sep.
Many genes are candidates for involvement in epileptic encephalopathy (EE) because one or a few possibly pathogenic variants have been found in patients, but insufficient genetic or functional evidence exists for a definite annotation.
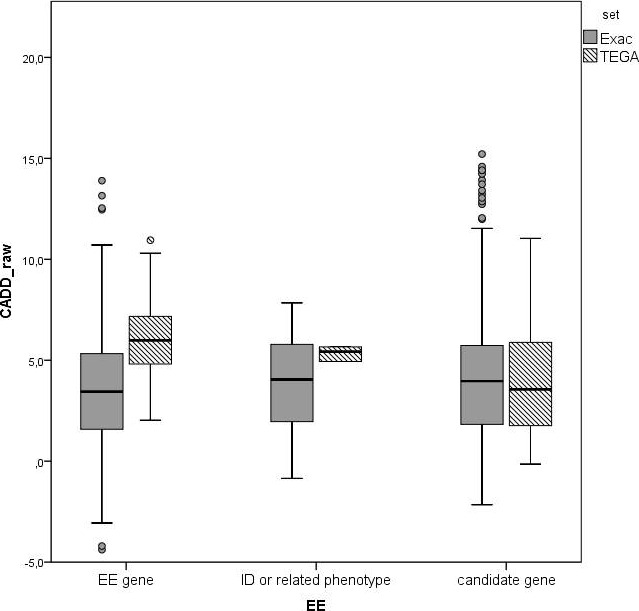
To increase the number of validated EE genes, we sequenced 26 known and 351 candidate genes for EE in 360 patients. Variants in 25 genes known to be involved in EE or related phenotypes were followed up in 41 patients. We prioritized the candidate genes, and followed up 31 variants in this prioritized subset of candidate genes.
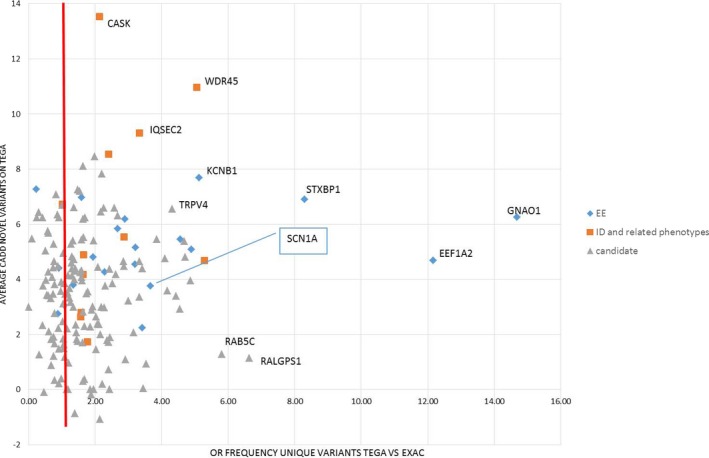
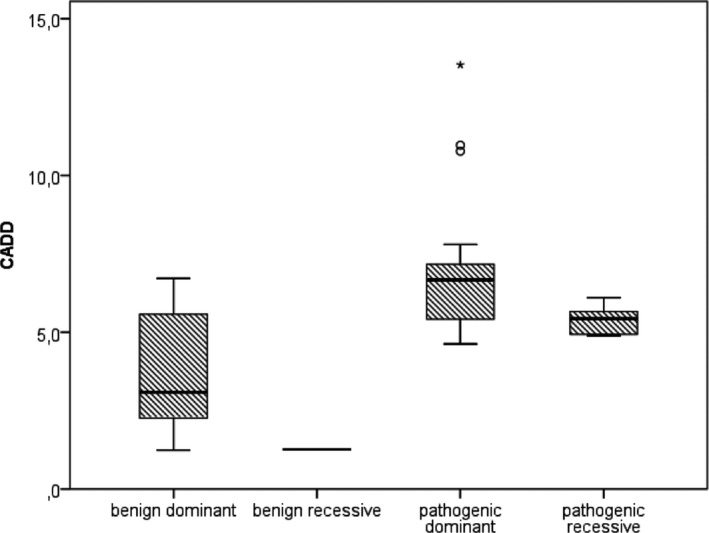
Twenty-nine genotypes in known genes for EE (19) or related diseases (10), dominant as well as recessive or X-linked, were classified as likely pathogenic variants. Among those, likely pathogenic de novo variants were found in EE genes that act dominantly, including the recently identified genes EEF1A2, KCNB1 and the X-linked gene IQSEC2. A de novo frameshift variant in candidate gene HNRNPU was the only de novo variant found among the followed-up candidate genes, and the patient's phenotype was similar to a few recent publications.
Mutations in genes described in OMIM as, for example, intellectual disability gene can lead to phenotypes that get classified as EE in the clinic. We confirmed existing literature reports that de novo loss-of-function HNRNPUmutations lead to severe developmental delay and febrile seizures in the first year of life.
许多基因都可能参与癫痫性脑病(EE),因为在患者中发现了一个或几个可能致病的变异,但缺乏足够的遗传或功能证据进行明确注释。
为了增加已验证的EE基因数量,我们对360例患者的26个已知EE基因和351个候选基因进行了测序。对41例患者中已知参与EE或相关表型的25个基因中的变异进行了随访。我们对候选基因进行了优先级排序,并对该优先级候选基因子集中的31个变异进行了随访。
EE已知基因(19个)或相关疾病(10个)中的29种基因型,包括显性、隐性或X连锁型,被分类为可能的致病变异。其中,在显性作用的EE基因中发现了可能的新生致病变异,包括最近鉴定的基因EEF1A2、KCNB1和X连锁基因IQSEC2。候选基因HNRNPU中的一个新生移码变异是随访的候选基因中唯一发现的新生变异,该患者的表型与最近的一些出版物报道相似。
《医学遗传学在线》(OMIM)中描述的基因,例如智力残疾基因的突变,可导致在临床上被归类为EE的表型。我们证实了现有文献报道,即新生的功能丧失性HNRNPU突变会导致出生后第一年严重的发育迟缓及热性惊厥。