Wyssenbach Ane, Quintela Tania, Llavero Francisco, Zugaza Jose L, Matute Carlos, Alberdi Elena
Departamento de Neurociencias, Universidad del País Vasco (UPV/EHU), 48940, Leioa, Spain.
Centro de Investigación en Red de Enfermedades Neurodegenerativas (CIBERNED), Leioa, Spain.
Aging Cell. 2016 Dec;15(6):1140-1152. doi: 10.1111/acel.12521. Epub 2016 Oct 5.
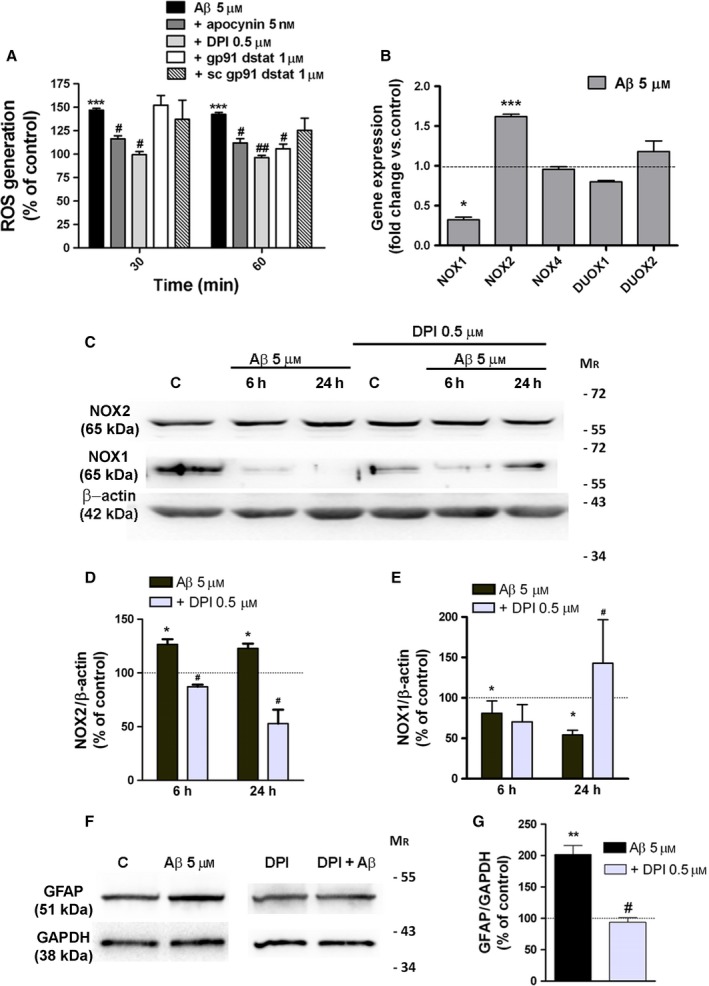
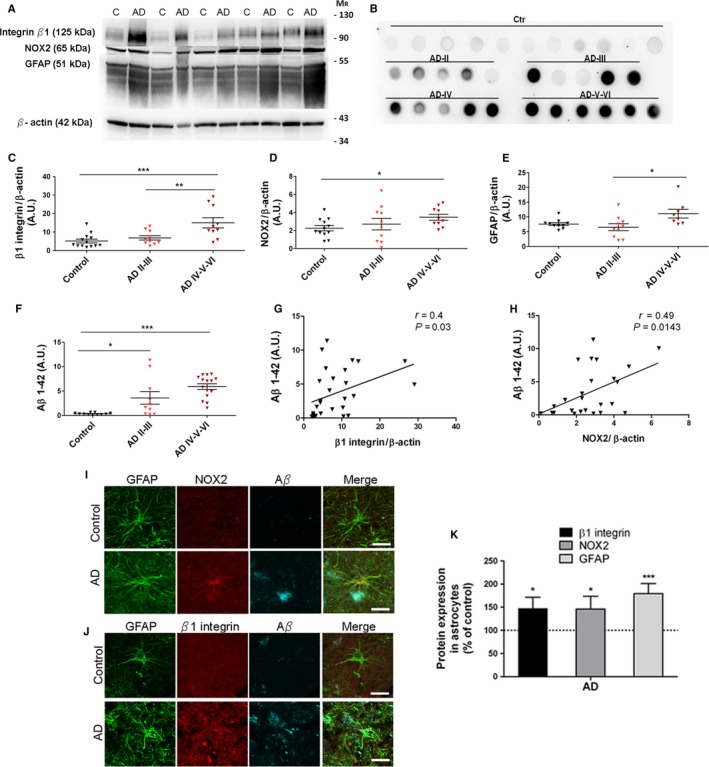
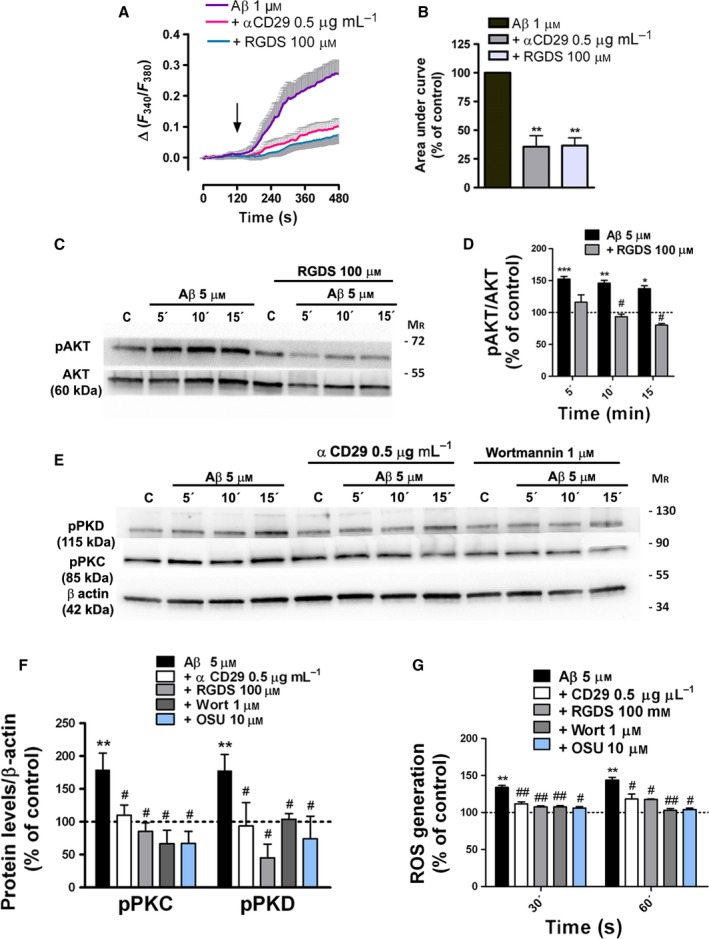
Astrogliosis is a hallmark of Alzheimer's disease (AD) and may constitute a primary pathogenic component of that disorder. Elucidation of signaling cascades inducing astrogliosis should help characterizing the function of astrocytes and identifying novel molecular targets to modulate AD progression. Here, we describe a novel mechanism by which soluble amyloid-β modulates β1-integrin activity and triggers NADPH oxidase (NOX)-dependent astrogliosis in vitro and in vivo. Amyloid-β oligomers activate a PI3K/classical PKC/Rac1/NOX pathway which is initiated by β1-integrin in cultured astrocytes. This mechanism promotes β1-integrin maturation, upregulation of NOX2 and of the glial fibrillary acidic protein (GFAP) in astrocytes in vitro and in hippocampal astrocytes in vivo. Notably, immunochemical analysis of the hippocampi of a triple-transgenic AD mouse model shows increased levels of GFAP, NOX2, and β1-integrin in reactive astrocytes which correlates with the amyloid β-oligomer load. Finally, analysis of these proteins in postmortem frontal cortex from different stages of AD (II to V/VI) and matched controls confirmed elevated expression of NOX2 and β1-integrin in that cortical region and specifically in reactive astrocytes, which was most prominent at advanced AD stages. Importantly, protein levels of NOX2 and β1-integrin were significantly associated with increased amyloid-β load in human samples. These data strongly suggest that astrogliosis in AD is caused by direct interaction of amyloid β oligomers with β1-integrin which in turn leads to enhancing β1-integrin and NOX2 activity via NOX-dependent mechanisms. These observations may be relevant to AD pathophysiology.
星形胶质细胞增生是阿尔茨海默病(AD)的一个标志,可能是该疾病的主要致病成分。阐明诱导星形胶质细胞增生的信号级联反应,应有助于明确星形胶质细胞的功能,并识别出调节AD进展的新分子靶点。在此,我们描述了一种新机制,即可溶性淀粉样β蛋白在体外和体内调节β1整合素活性并触发NADPH氧化酶(NOX)依赖性星形胶质细胞增生。淀粉样β寡聚体激活PI3K/经典蛋白激酶C/Rac1/NOX信号通路,该通路由培养的星形胶质细胞中的β1整合素启动。这种机制促进β1整合素成熟,在体外培养的星形胶质细胞以及体内海马星形胶质细胞中上调NOX2和胶质纤维酸性蛋白(GFAP)。值得注意的是,对三转基因AD小鼠模型海马的免疫化学分析显示,反应性星形胶质细胞中GFAP、NOX2和β1整合素水平升高,这与淀粉样β寡聚体负荷相关。最后,对来自AD不同阶段(II至V/VI)的死后额叶皮质及匹配对照中这些蛋白的分析证实,该皮质区域尤其是反应性星形胶质细胞中NOX2和β1整合素表达升高,在AD晚期最为显著。重要的是,在人类样本中,NOX2和β1整合素的蛋白水平与淀粉样β负荷增加显著相关。这些数据强烈表明,AD中的星形胶质细胞增生是由淀粉样β寡聚体与β1整合素的直接相互作用引起的,进而通过NOX依赖性机制增强β1整合素和NOX2活性。这些观察结果可能与AD的病理生理学相关。