Kitaura Kazutaka, Shini Tadasu, Matsutani Takaji, Suzuki Ryuji
Repertoire Genesis Incorporation, 104 Saito-Bioincubator, 7-7-15, Saito-asagi, Ibaraki, Osaka, 567-0085, Japan.
Department of Rheumatology and Clinical Immunology, Clinical Research Center for Rheumatology and Allergy, Sagamihara National Hospital, National Hospital Organization, Sagamihara, Japan.
BMC Immunol. 2016 Oct 11;17(1):38. doi: 10.1186/s12865-016-0177-5.
High-throughput sequencing of T cell receptor (TCR) genes is a powerful tool for analyses of antigen specificity, clonality and diversity of T lymphocytes. Here, we developed a new TCR repertoire analysis method using 454 DNA sequencing technology in combination with an adaptor-ligation mediated polymerase chain reaction (PCR). This method allows the amplification of all TCR genes without PCR bias. To compare gene usage, diversity and similarity of expressed TCR repertoires among individuals, we conducted next-generation sequencing (NGS) of TRA and TRB genes in peripheral blood mononuclear cells from 20 healthy human individuals.
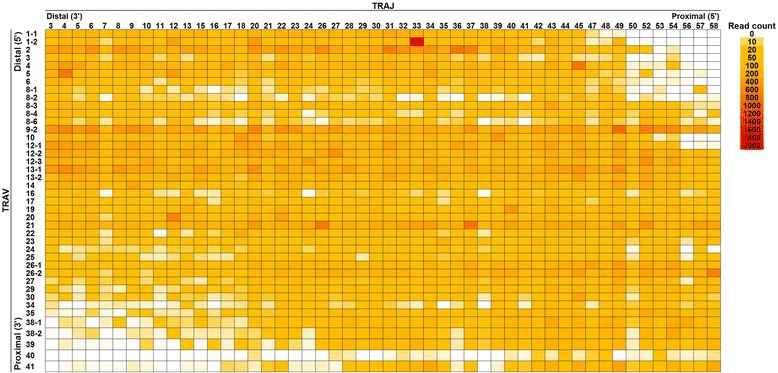
From a total of 267,037 sequence reads from 20 individuals, 149,216 unique sequence reads were identified. Preferential usage of several V and J genes were observed while some recombinations of TRAV with TRAJ appeared to be restricted. The extent of TCR diversity was not significantly different between TRA and TRB, while TRA repertoires were more similar between individuals than TRB repertoires were. The interindividual similarity of TRA depended largely on the frequent presence of shared TCRs among two or more individuals. A publicly available TRA had a near-germline TCR with a shorter CDR3. Notably, shared TRA sequences, especially those shared among a large number of individuals', often contained TCRα related with invariant TCRα derived from invariant natural killer T cells and mucosal-associated invariant T cells.
These results suggest that retrieval of shared TCRs by NGS would be useful for the identification of potential new invariant TCRα chains. This NGS method will enable the comprehensive quantitative analysis of TCR repertoires at a clonal level.
T细胞受体(TCR)基因的高通量测序是分析T淋巴细胞抗原特异性、克隆性和多样性的有力工具。在此,我们开发了一种新的TCR库分析方法,该方法将454 DNA测序技术与衔接子连接介导的聚合酶链反应(PCR)相结合。此方法能够无PCR偏差地扩增所有TCR基因。为了比较个体间表达的TCR库的基因使用情况、多样性和相似性,我们对20名健康人类个体外周血单个核细胞中的TRA和TRB基因进行了新一代测序(NGS)。
从20名个体的总共267,037条序列读数中,鉴定出149,216条独特的序列读数。观察到几个V和J基因的优先使用情况,同时TRAV与TRAJ的一些重组似乎受到限制。TRA和TRB之间TCR多样性的程度没有显著差异,而个体间TRA库比TRB库更相似。TRA的个体间相似性在很大程度上取决于两个或更多个体之间频繁存在共享的TCR。一个公开可用的TRA具有近乎胚系的TCR,其CDR3较短。值得注意的是,共享的TRA序列,尤其是那些在大量个体之间共享的序列,通常包含与源自不变自然杀伤T细胞和黏膜相关不变T细胞的不变TCRα相关的TCRα。
这些结果表明,通过NGS检索共享的TCR对于鉴定潜在的新的不变TCRα链将是有用的。这种NGS方法将能够在克隆水平上对TCR库进行全面的定量分析。