Shen Yu-Min
Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, TX 75390-8852 USA.
Thromb J. 2016 Oct 4;14(Suppl 1):19. doi: 10.1186/s12959-016-0114-0. eCollection 2016.
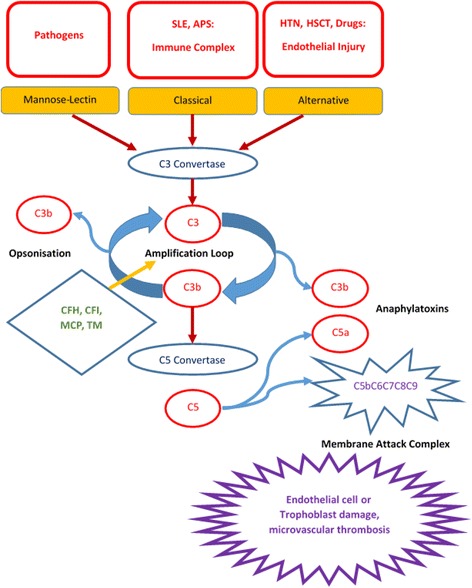
Atypical hemolytic uremic syndrome (aHUS) is a rare genetic disorder caused by defective complement regulation resulting in thrombotic microangiopathy (TMA). Patients can present as children or adults. The syndrome consists of hemolytic anemia with schistocytosis, thrombocytopenia, significant renal damage, and/or other organ system dysfunction(s). Patients with aHUS may succumb to the complications of the disease with the very first manifestation; surviving patients often suffer from progressive organ dysfunction with significant morbidity and mortality despite plasma infusion or plasma exchange. Eculizumab, a humanized monoclonal antibody to C5, was approved for treatment of aHUS in 2011. This is an expensive but highly effective therapy changing the lives and improving the outcome of patients with aHUS. Making timely and accurate diagnosis of aHUS can be life-saving if eculizumab treatment is begun promptly. Finding a genetic mutation in a complement regulatory protein is diagnostic with the appropriate clinical syndrome, but at least 30 % of patients do not have defined or reported mutations. Thus the diagnosis rests on the clinical acumen of the physician. However, the clinical manifestations of aHUS are shared by other etiologies of thrombotic microangiopathy. While laboratory finding of undetectable ADAMTS13 activity defines TTP, distinguishing aHUS from the other causes of TMA remains an art. In addition, aHUS can be unmasked by conditions with enhanced complement activation, such as systemic lupus erythematosus, pregnancy, malignant hypertension, and hematopoietic stem cell transplantation. Thus if TMA occurs in the setting of enhanced complement activation, one must consider aHUS as an underlying etiology, especially if treatment of the condition does not resolve the TMA.
非典型溶血性尿毒症综合征(aHUS)是一种罕见的遗传性疾病,由补体调节缺陷导致血栓性微血管病(TMA)。患者可在儿童期或成年期发病。该综合征包括伴有破碎红细胞的溶血性贫血、血小板减少、严重肾损害和/或其他器官系统功能障碍。aHUS患者可能在首次出现症状时就死于该病的并发症;存活患者尽管接受了血浆输注或血浆置换,仍常遭受进行性器官功能障碍,发病率和死亡率都很高。依库珠单抗是一种针对C5的人源化单克隆抗体,于2011年被批准用于治疗aHUS。这是一种昂贵但非常有效的疗法,改变了aHUS患者的生活并改善了其预后。如果能及时开始依库珠单抗治疗,及时准确地诊断aHUS可挽救生命。在补体调节蛋白中发现基因突变并伴有相应的临床综合征即可确诊,但至少30%的患者没有明确或报道的突变。因此,诊断依赖于医生的临床敏锐度。然而,aHUS的临床表现与血栓性微血管病的其他病因有重叠。虽然实验室检查发现ADAMTS13活性无法检测可诊断为血栓性血小板减少性紫癜(TTP),但将aHUS与TMA的其他病因区分开来仍然是一门艺术。此外,aHUS可在补体激活增强的情况下被诱发,如系统性红斑狼疮、妊娠、恶性高血压和造血干细胞移植。因此,如果在补体激活增强的情况下发生TMA,必须考虑aHUS作为潜在病因,尤其是在针对该病症的治疗未能解决TMA的情况下。