Karim Sajjad, NourEldin Hend Fakhri, Abusamra Heba, Salem Nada, Alhathli Elham, Dudley Joel, Sanderford Max, Scheinfeldt Laura B, Chaudhary Adeel G, Al-Qahtani Mohammed H, Kumar Sudhir
Center for Excellence in Genome Medicine and Research, King Abdulaziz University, Jeddah, Saudi Arabia.
Department of Genetics and Genomic Sciences, Mount Sinai School of Medicine, New York, NY, 10029, USA.
BMC Genomics. 2016 Oct 17;17(Suppl 9):770. doi: 10.1186/s12864-016-3088-1.
Genome-wide association studies (GWAS) have become a mainstay of biological research concerned with discovering genetic variation linked to phenotypic traits and diseases. Both discrete and continuous traits can be analyzed in GWAS to discover associations between single nucleotide polymorphisms (SNPs) and traits of interest. Associations are typically determined by estimating the significance of the statistical relationship between genetic loci and the given trait. However, the prioritization of bona fide, reproducible genetic associations from GWAS results remains a central challenge in identifying genomic loci underlying common complex diseases. Evolutionary-aware meta-analysis of the growing GWAS literature is one way to address this challenge and to advance from association to causation in the discovery of genotype-phenotype relationships.
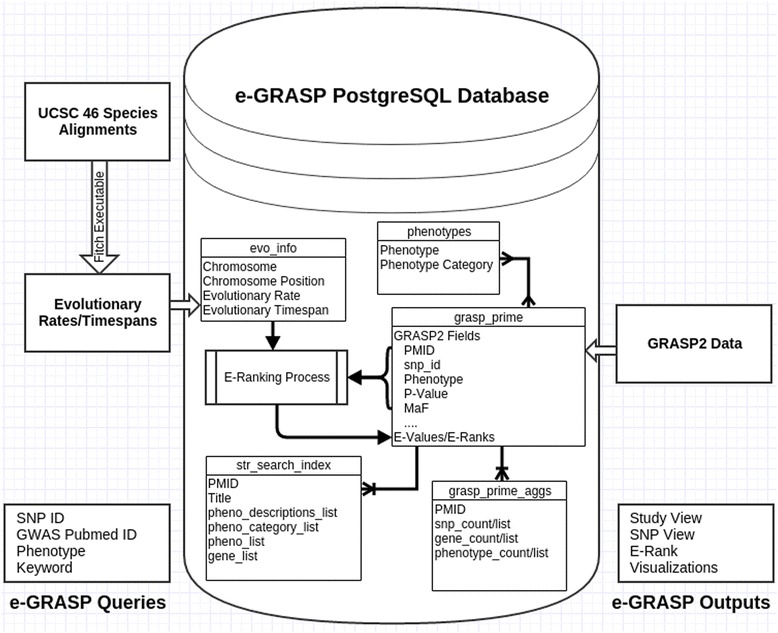
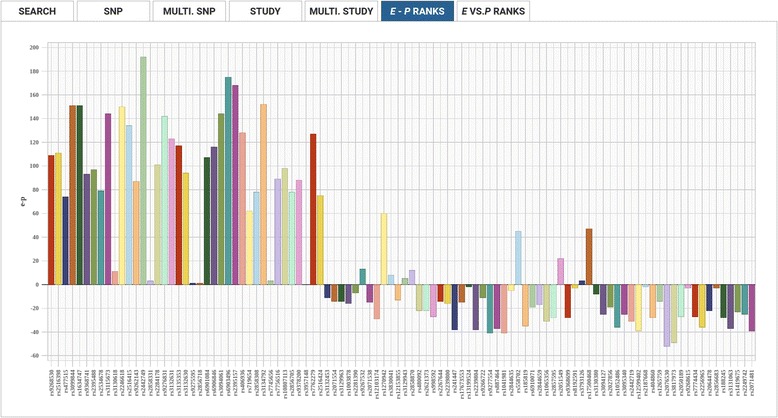
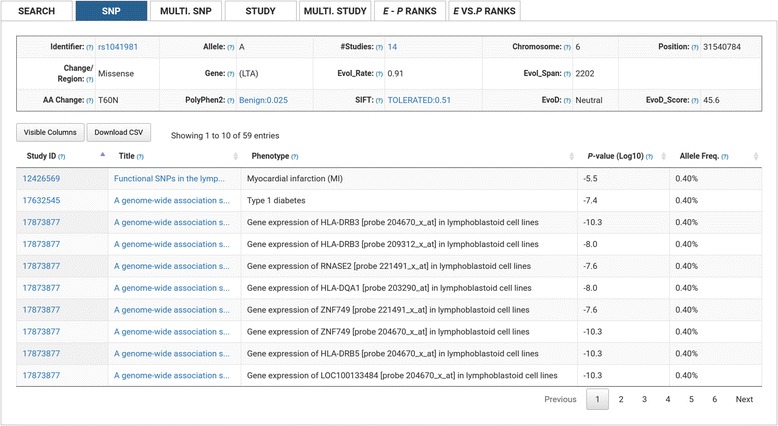
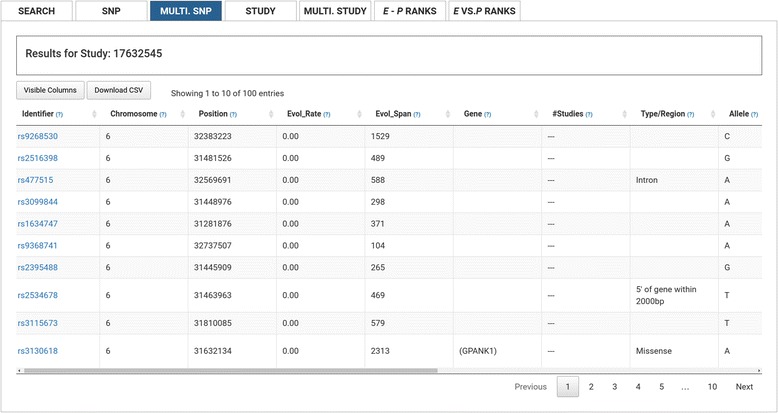
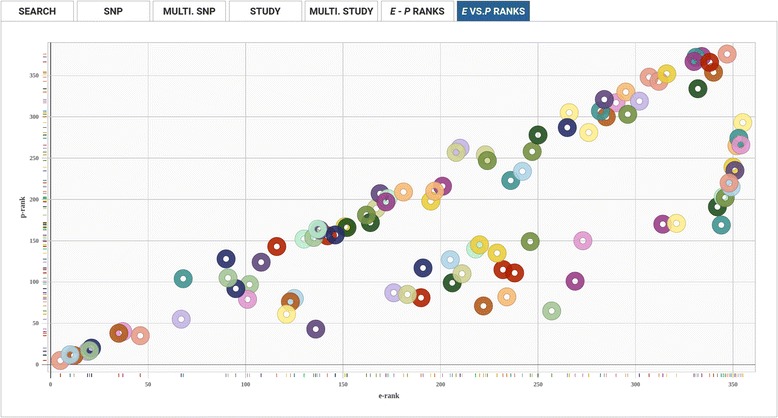
We have created an evolutionary GWAS resource to enable in-depth query and exploration of published GWAS results. This resource uses the publically available GWAS results annotated in the GRASP2 database. The GRASP2 database includes results from 2082 studies, 177 broad phenotype categories, and ~8.87 million SNP-phenotype associations. For each SNP in e-GRASP, we present information from the GRASP2 database for convenience as well as evolutionary information (e.g., rate and timespan). Users can, therefore, identify not only SNPs with highly significant phenotype-association P-values, but also SNPs that are highly replicated and/or occur at evolutionarily conserved sites that are likely to be functionally important. Additionally, we provide an evolutionary-adjusted SNP association ranking (E-rank) that uses cross-species evolutionary conservation scores and population allele frequencies to transform P-values in an effort to enhance the discovery of SNPs with a greater probability of biologically meaningful disease associations.
By adding an evolutionary dimension to the GWAS results available in the GRASP2 database, our e-GRASP resource will enable a more effective exploration of SNPs not only by the statistical significance of trait associations, but also by the number of studies in which associations have been replicated, and the evolutionary context of the associated mutations. Therefore, e-GRASP will be a valuable resource for aiding researchers in the identification of bona fide, reproducible genetic associations from GWAS results. This resource is freely available at http://www.mypeg.info/egrasp .
全基因组关联研究(GWAS)已成为生物学研究的主要手段,用于发现与表型特征和疾病相关的基因变异。离散和连续性状均可在GWAS中进行分析,以发现单核苷酸多态性(SNP)与感兴趣性状之间的关联。关联通常通过估计基因座与给定性状之间统计关系的显著性来确定。然而,从GWAS结果中确定真正的、可重复的基因关联的优先级仍然是识别常见复杂疾病潜在基因组位点的核心挑战。对不断增长的GWAS文献进行进化感知的荟萃分析是应对这一挑战并在发现基因型-表型关系方面从关联推进到因果关系的一种方法。
我们创建了一个进化GWAS资源,以实现对已发表GWAS结果的深入查询和探索。该资源使用GRASP2数据库中注释的公开可用GWAS结果。GRASP2数据库包含来自2082项研究、177个广泛表型类别和约887万个SNP-表型关联的结果。为了方便起见,对于电子GRASP中的每个SNP,我们展示了来自GRASP2数据库的信息以及进化信息(例如,速率和时间跨度)。因此,用户不仅可以识别具有高度显著表型关联P值的SNP,还可以识别高度重复和/或出现在可能具有功能重要性的进化保守位点的SNP。此外,我们提供了一种进化调整后的SNP关联排名(E-rank),它使用跨物种进化保守分数和群体等位基因频率来转换P值,以努力增强对具有更大生物学意义疾病关联可能性的SNP的发现。
通过在GRASP2数据库中可用的GWAS结果中增加一个进化维度,我们的电子GRASP资源将不仅能够通过性状关联的统计显著性,还能通过关联已被重复的研究数量以及相关突变的进化背景,更有效地探索SNP。因此,电子GRASP将成为帮助研究人员从GWAS结果中识别真正的、可重复的基因关联的宝贵资源。该资源可在http://www.mypeg.info/egrasp免费获取。