Hoppmann Anselm S, Schlosser Pascal, Backofen Rolf, Lausch Ekkehart, Köttgen Anna
Dept. of Pediatric Genetics, Medical Center - University of Freiburg, Faculty of Medicine, University of Freiburg, Freiburg, Germany.
Division of Genetic Epidemiology, Institute for Medical Biometry and Statistics, Medical Center - University of Freiburg, Faculty of Medicine, University of Freiburg, Freiburg, Germany.
PLoS One. 2016 Sep 9;11(9):e0162466. doi: 10.1371/journal.pone.0162466. eCollection 2016.
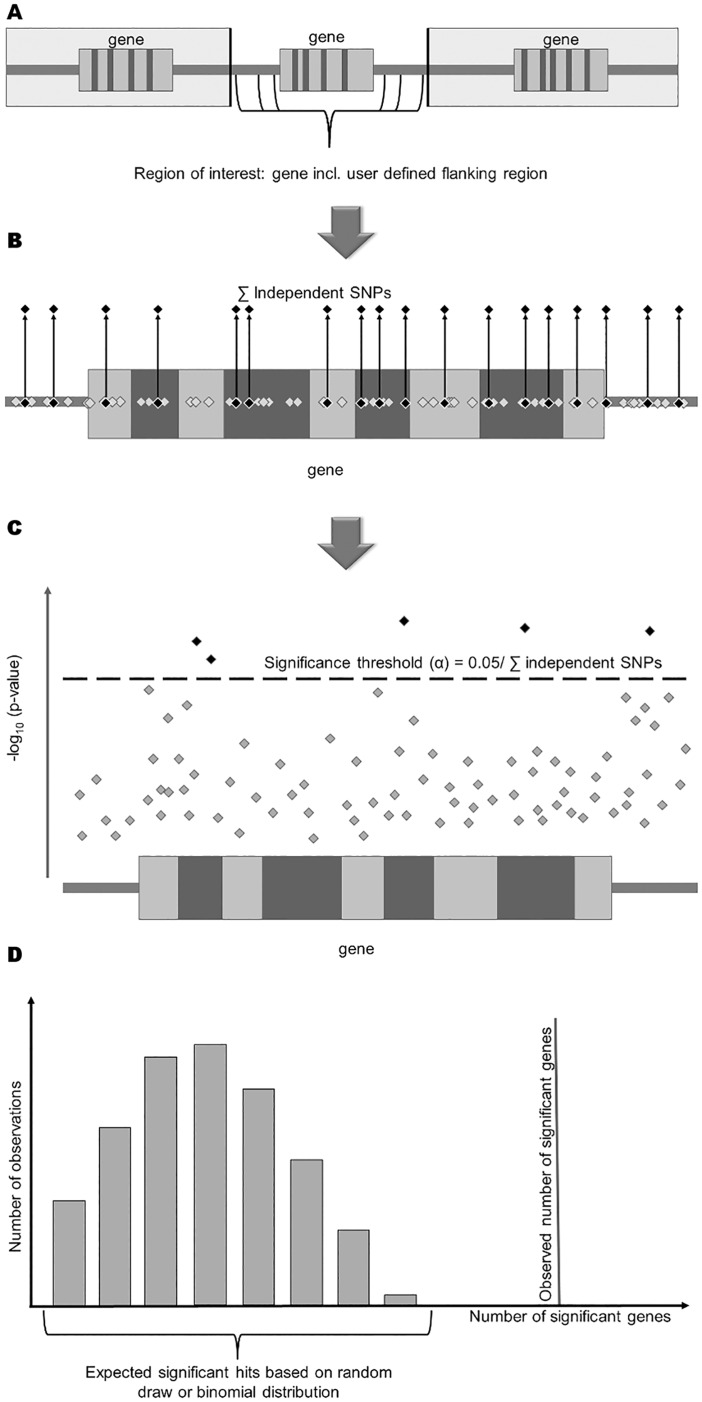
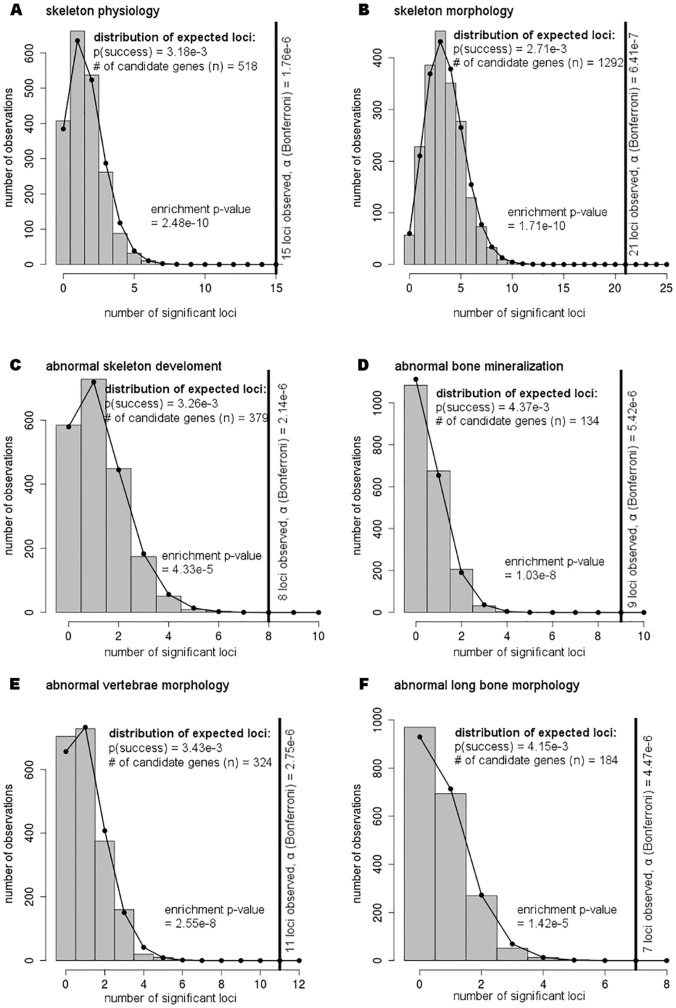
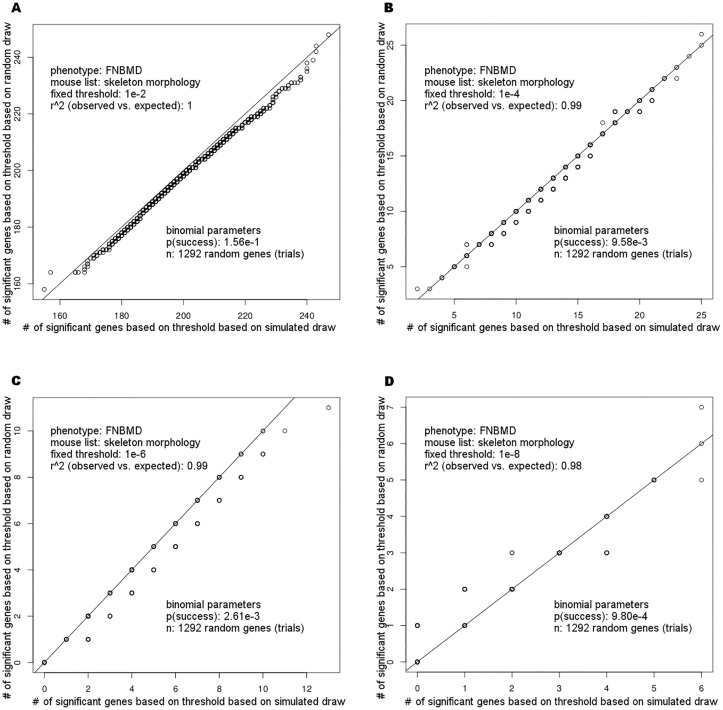
Genome-wide association studies (GWAS) evaluate associations between genetic variants and a trait or disease of interest free of prior biological hypotheses. GWAS require stringent correction for multiple testing, with genome-wide significance typically defined as association p-value <5*10-8. This study presents a new tool that uses external information about genes to prioritize SNP associations (GenToS). For a given list of candidate genes, GenToS calculates an appropriate statistical significance threshold and then searches for trait-associated variants in summary statistics from human GWAS. It thereby allows for identifying trait-associated genetic variants that do not meet genome-wide significance. The program additionally tests for enrichment of significant candidate gene associations in the human GWAS data compared to the number expected by chance. As proof of principle, this report used external information from a comprehensive resource of genetically manipulated and systematically phenotyped mice. Based on selected murine phenotypes for which human GWAS data for corresponding traits were publicly available, several candidate gene input lists were derived. Using GenToS for the investigation of candidate genes underlying murine skeletal phenotypes in data from a large human discovery GWAS meta-analysis of bone mineral density resulted in the identification of significantly associated variants in 29 genes. Index variants in 28 of these loci were subsequently replicated in an independent GWAS replication step, highlighting that they are true positive associations. One signal, COL11A1, has not been discovered through GWAS so far and represents a novel human candidate gene for altered bone mineral density. The number of observed genes that contained significant SNP associations in human GWAS based on murine candidate gene input lists was much greater than the number expected by chance across several complex human traits (enrichment p-value as low as 10-10). GenToS can be used with any candidate gene list, any GWAS summary file, runs on a desktop computer and is freely available.
全基因组关联研究(GWAS)在没有先验生物学假设的情况下评估基因变异与感兴趣的性状或疾病之间的关联。GWAS需要对多重检验进行严格校正,全基因组显著性通常定义为关联p值<5×10⁻⁸。本研究提出了一种新工具,该工具利用有关基因的外部信息对单核苷酸多态性(SNP)关联进行优先级排序(GenToS)。对于给定的候选基因列表,GenToS计算适当的统计显著性阈值,然后在人类GWAS的汇总统计数据中搜索与性状相关的变异。因此,它能够识别未达到全基因组显著性的与性状相关的基因变异。该程序还会在人类GWAS数据中测试显著候选基因关联的富集情况,并与随机预期的数量进行比较。作为原理验证,本报告使用了来自经过基因操作和系统表型分析的小鼠综合资源的外部信息。基于选定的小鼠表型(相应性状的人类GWAS数据已公开),得出了几个候选基因输入列表。在一项关于骨密度的大型人类发现GWAS荟萃分析的数据中,使用GenToS研究小鼠骨骼表型潜在的候选基因,结果在29个基因中鉴定出显著相关的变异。随后,在一个独立的GWAS复制步骤中复制了其中28个位点的索引变异,突出表明它们是真正的阳性关联。一个信号,即COL11A1,迄今为止尚未通过GWAS发现,它代表了一个新的骨密度改变的人类候选基因。基于小鼠候选基因输入列表,在人类GWAS中观察到的包含显著SNP关联的基因数量,远大于在几个复杂人类性状中随机预期的数量(富集p值低至10⁻¹⁰)。GenToS可与任何候选基因列表、任何GWAS汇总文件一起使用,在台式计算机上运行,并且可免费获取。