Kratsch Christina, Klingen Thorsten R, Mümken Linda, Steinbrück Lars, McHardy Alice C
Department for Algorithmic Bioinformatics, Heinrich Heine University, Düsseldorf, Germany and.
Department for Algorithmic Bioinformatics, Heinrich Heine University, Düsseldorf, Germany and; Department for Computational Biology of Infection Research, Helmholtz Center for Infection Research, Braunschweig, Germany.
Virus Evol. 2016 Feb 14;2(1):vev025. doi: 10.1093/ve/vev025. eCollection 2016 Jan.
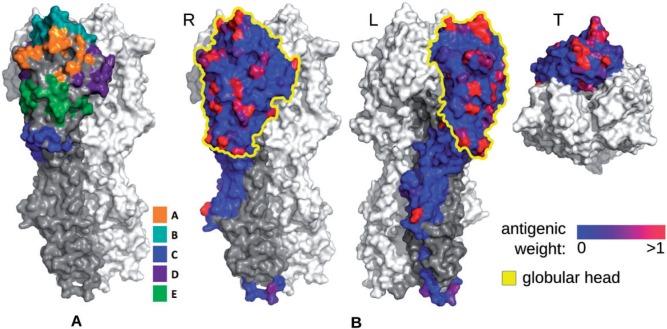
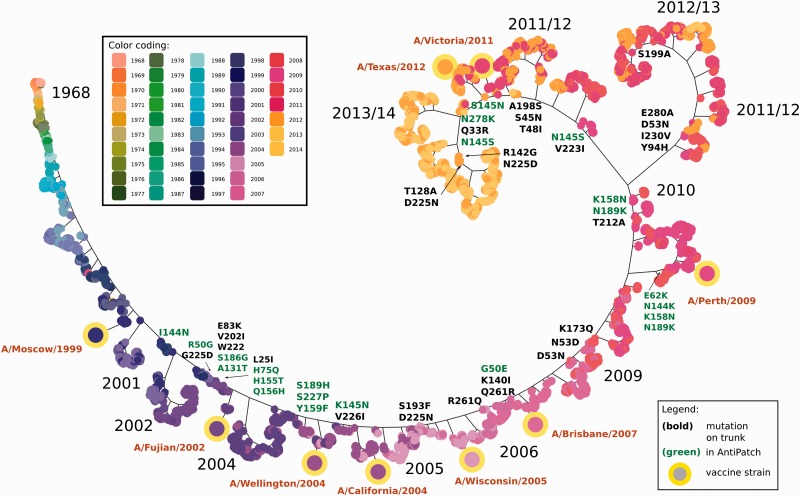
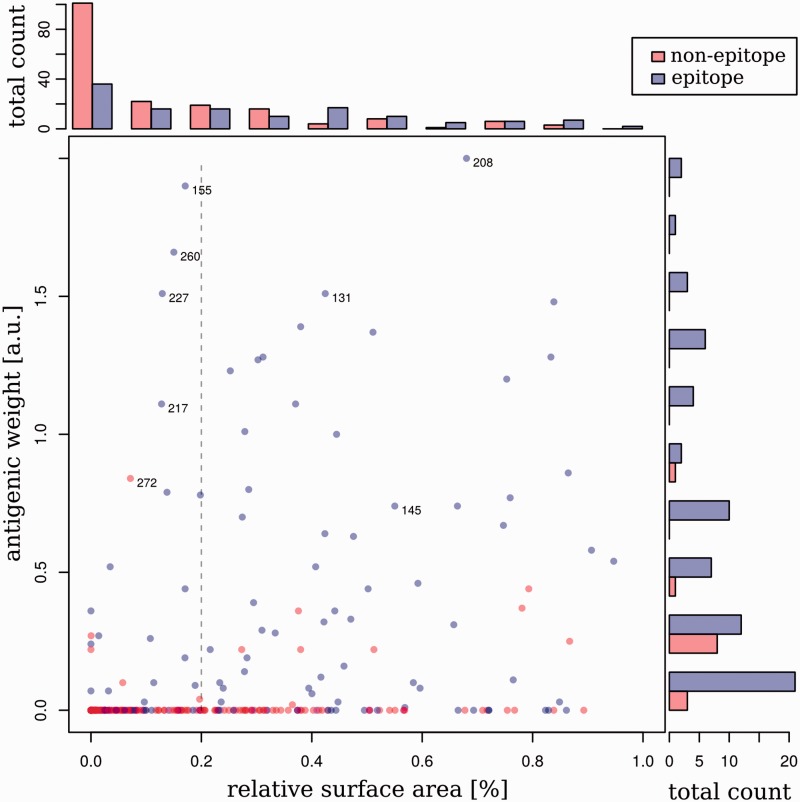
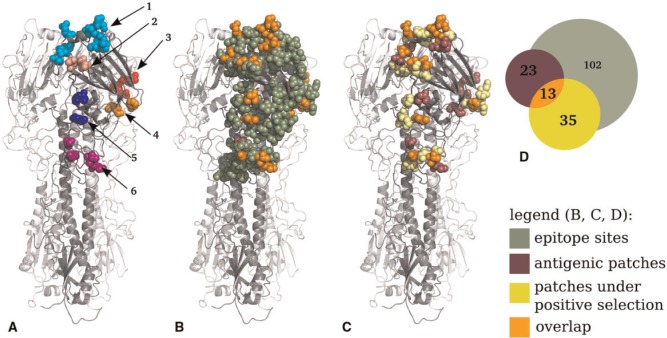
Human influenza viruses are rapidly evolving RNA viruses that cause short-term respiratory infections with substantial morbidity and mortality in annual epidemics. Uncovering the general principles of viral coevolution with human hosts is important for pathogen surveillance and vaccine design. Protein regions are an appropriate model for the interactions between two macromolecules, but the currently used epitope definition for the major antigen of influenza viruses, namely hemagglutinin, is very broad. Here, we combined genetic, evolutionary, antigenic, and structural information to determine the most relevant regions of the hemagglutinin of human influenza A/H3N2 viruses for interaction with human immunoglobulins. We estimated the antigenic weights of amino acid changes at individual sites from hemagglutination inhibition data using antigenic tree inference followed by spatial clustering of antigenicity-altering protein sites on the protein structure. This approach determined six relevant areas (patches) for antigenic variation that had a key role in the past antigenic evolution of the viruses. Previous transitions between successive predominating antigenic types of H3N2 viruses always included amino acid changes in either the first or second antigenic patch. Interestingly, there was only partial overlap between the antigenic patches and the patches under strong positive selection. Therefore, besides alterations of antigenicity, other interactions with the host may shape the evolution of human influenza A/H3N2 viruses.
人类流感病毒是快速进化的RNA病毒,每年都会引发短期呼吸道感染,导致大量发病和死亡。揭示病毒与人类宿主共同进化的一般原则对于病原体监测和疫苗设计至关重要。蛋白质区域是两种大分子之间相互作用的合适模型,但目前用于流感病毒主要抗原(即血凝素)的表位定义非常宽泛。在这里,我们结合遗传、进化、抗原和结构信息,以确定甲型H3N2流感病毒血凝素与人类免疫球蛋白相互作用的最相关区域。我们使用抗原树推断从血凝抑制数据估计单个位点氨基酸变化的抗原权重,随后对蛋白质结构上改变抗原性的蛋白质位点进行空间聚类。这种方法确定了六个与抗原变异相关的区域(斑块),它们在病毒过去的抗原进化中起关键作用。H3N2病毒连续占主导地位的抗原类型之间的先前转变总是包括第一或第二个抗原斑块中的氨基酸变化。有趣的是,抗原斑块与受到强烈正选择的斑块之间只有部分重叠。因此,除了抗原性改变外,与宿主的其他相互作用可能会影响甲型H3N2流感病毒的进化。